GenoPred Pipeline - Benchmark
In this document we will benchmark the time taken and max memory for each part of the GenoPred pipeline. We are using the benchmark results from the snakemake benchmark functionality. We will record the time taken to run the pipeline restricted to chromosome 22 to save time, and then results can be extrapolated.
We will test the pipeline using 10 GWAS, 10 external scoring files, 3 target samples with sample sizes 100, 1000 and 10000, with 10M SNPs originally. The number of SNPs in external scoring files can vary a lot, so we will use scoring files based on methods restricted to hapmap3 variants.
Prepare input data
# Create directory for inputs
mkdir -p /users/k1806347/oliverpainfel/test/genopred_benchmark/input_dataTarget data
Show code
# Read in previous population probabilities
library(data.table)
ukb_pop <- fread('/scratch/prj/ukbiobank/usr/ollie_pain/ReQC/defining_ancestry/UKBB.Ancestry.model_pred')
# Insert project specific IDs
fam <-fread('/scratch/prj/ukbiobank/ukb82087/genotyped/ukb82087_binary_pre_qc.fam')
fam$row<-1:nrow(fam)
ukb_pop <- merge(ukb_pop, fam[,c('V1','row'), with=F], by.x='FID',by.y='row')
# Identify subset with EUR probability > 0.995
ukb_pop_eur <- ukb_pop[ukb_pop$EUR > 0.995,]
ukb_pop_eur <- data.table(FID = ukb_pop_eur$V1,
IID = ukb_pop_eur$V1)
# Remove withdrawals
psam <- fread('/scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.psam')
names(psam)[1:2]<-c('FID','IID')
ukb_pop_eur <- ukb_pop_eur[ukb_pop_eur$FID %in% psam$FID,]
# Save keep file listing 100, 1000, and 10000 individuals
write.table(ukb_pop_eur[1:100,], '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_1.keep', col.names = F, row.names = F, quote = F)
write.table(ukb_pop_eur[1:1000,], '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_2.keep', col.names = F, row.names = F, quote = F)
write.table(ukb_pop_eur[1:10000,], '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_3.keep', col.names = F, row.names = F, quote = F)# Create symlinks to the pgen files resitricted to INFO 0.4 and MAF 0.01
ln -s /scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.psam /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22.psam
ln -s /scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.bgen.bgi /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22.bgen.bgi
ln -s /scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.pvar /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22.pvar
ln -s /scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.pgen /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22.pgen
ln -s /scratch/prj/ukbiobank/ukb82087/imputed/ukb82087_imp_chr22_MAF1_INFO4_v1.sample /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22.sample
# Subset the plink2 format files
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_3.keep \
--make-pgen \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_2.keep \
--make-pgen \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_1.keep \
--make-pgen \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.chr22
# Convert to bgen format
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_3.keep \
--export bgen-1.3 \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_2.keep \
--export bgen-1.3 \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22
plink2 --pfile /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.chr22 \
--keep /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb_eur_subset_1.keep \
--export bgen-1.3 \
--threads 1 \
--out /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.chr22
# Index the bgen files
/users/k1806347/oliverpainfel/Software/QCTOOL/bgen.tgz/build/apps/bgenix -index -g /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.chr22.bgen
/users/k1806347/oliverpainfel/Software/QCTOOL/bgen.tgz/build/apps/bgenix -index -g /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22.bgen
/users/k1806347/oliverpainfel/Software/QCTOOL/bgen.tgz/build/apps/bgenix -index -g /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22.bgen
# Rename sample file
mv /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.chr22.sample /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1.sample
mv /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.chr22.sample /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2.sample
mv /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.chr22.sample /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3.sample# Create a target_list
target_list <- data.frame(
name = c('subset_1_plink2', 'subset_2_plink2', 'subset_3_plink2', 'subset_1_bgen', 'subset_2_bgen', 'subset_3_bgen'),
path = c('/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_1', '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_2', '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/ukb.subset_3'),
type = c('plink2','plink2','plink2','bgen','bgen','bgen'),
indiv_report = c(T, F, F, F, F, F)
)
write.table(target_list, '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/target_list.txt', col.names=T, row.names=F, quote=F)GWAS data
Show code
# Create GWAS list
gwas_list <-data.frame(name = c('DEPR06','COLL01','BODY04','HEIG03','DIAB05','COAD01','SCLE03','RHEU02','BRCA01','PRCA01'),
path = NA,
population = 'EUR',
n = NA,
sampling = c(0.2698322, NA, NA, NA, 0.1675544, 0.3298934, 0.3599529, 0.2463970, 0.5371324, 0.5643190),
prevalence = c(0.15, NA, NA, NA, 0.05, 0.03, 0.00164, 0.005, 0.125, 0.125),
mean = c(NA,0,0,0,NA,NA,NA,NA,NA,NA),
sd = c(NA,1,1,1,NA,NA,NA,NA,NA,NA),
label = c('Major Depression','Intelligence','BMI','Height','T2D','CAD','MultiScler','RheuArth','Breast Cancer','Prostate Cancer'))
gwas_list$path <-paste0('/scratch/prj/gwas_sumstats/cleaned/', gwas_list$name,'.gz')
gwas_list$label<-paste0("\"", gwas_list$label, "\"")
write.table(gwas_list, '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/gwas_list.txt', col.names=T, row.names=F, quote=F)External score file
Show code
# Create a score list
score_list <- data.frame(name=c('PGS003980','PGS003981','PGS003982','PGS003983','PGS003984'),
path=NA,
label=c('body mass index','inflammatory bowel disease','type 2 diabetes mellitus','breast carcinoma','stroke'))
score_list$label<-paste0("\"", score_list$label, "\"")
write.table(score_list, '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/score_list.txt', col.names=T, row.names=F, quote=F)Configfile
Show code
config <- c(
"outdir: /users/k1806347/oliverpainfel/test/genopred_benchmark/output/ncores_1",
"config_file: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores1.yaml",
"gwas_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/gwas_list.txt",
"target_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/target_list.txt",
"score_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/score_list.txt",
"pgs_methods: ['ptclump','dbslmm','prscs','sbayesr','lassosum','ldpred2','megaprs']",
"testing: chr22",
"cores_prep_pgs: 1",
"cores_target_pgs: 1",
"cores_impute_23andme: 1",
"cores_outlier_detection: 1"
)
write.table(config, '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores1.yaml', col.names=F, row.names=F, quote=F)
config <- c(
"outdir: /users/k1806347/oliverpainfel/test/genopred_benchmark/output/ncores_5",
"config_file: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores5.yaml",
"gwas_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/gwas_list.txt",
"target_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/target_list.txt",
"score_list: /users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/score_list.txt",
"pgs_methods: ['ptclump','dbslmm','prscs','sbayesr','lassosum','ldpred2','megaprs']",
"testing: chr22",
"cores_prep_pgs: 5",
"cores_target_pgs: 5",
"cores_impute_23andme: 5",
"cores_outlier_detection: 5"
)
write.table(config, '/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores5.yaml', col.names=F, row.names=F, quote=F)Run pipeline
git describe --tags
#v2.2.5-13-g3771778
# N cores = 1
snakemake --profile slurm -n --configfile=/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores1.yaml --use-conda output_all pc_projection outlier_detection
# N cores = 5
snakemake --profile slurm -n --configfile=/users/k1806347/oliverpainfel/test/genopred_benchmark/input_data/config_ncores5.yaml --use-conda output_all pc_projection outlier_detection
Collate results
Show code
library(data.table)
library(ggplot2)
library(cowplot)
runs <- c('ncores_1','ncores_5')
bm_dat_all <- NULL
for (run_i in runs) {
# Identify benchmark files
bm_files_i <-
paste0(
'/users/k1806347/oliverpainfel/test/genopred_benchmark/output/',
run_i,
'/reference/benchmarks/',
list.files(
paste0(
'/users/k1806347/oliverpainfel/test/genopred_benchmark/output/',
run_i,
'/reference/benchmarks'
)
)
)
# Read in benchmark files
bm_dat_i <- do.call(rbind, lapply(bm_files_i, function(file) {
tmp <- fread(file)
tmp$file <- basename(file)
return(tmp)
}))
bm_dat_i$ncores <- as.numeric(gsub('ncores_','',run_i))
bm_dat_all<-rbind(bm_dat_all, bm_dat_i)
}
# Read in benchmark data that is configuration independent
bm_files_i <-
paste0(
'resources/data/benchmarks/',
list.files('resources/data/benchmarks')
)
# Read in benchmark files
bm_dat_i <- do.call(rbind, lapply(bm_files_i, function(file) {
tmp <- fread(file)
tmp$file <- basename(file)
return(tmp)
}))
bm_dat_i$ncores <- 1
bm_dat_all<-rbind(bm_dat_all, bm_dat_i)
# Create rule, n, pgs_method and type columns
bm_dat_all$rule <- gsub('-.*','',bm_dat_all$file)
bm_dat_all$n <- NA
bm_dat_all$type <- NA
bm_dat_all$pgs_method <- NA
bm_dat_all$n[grepl('subset_1', bm_dat_all$file)] <- 100
bm_dat_all$n[grepl('subset_2', bm_dat_all$file)] <- 1000
bm_dat_all$n[grepl('subset_3', bm_dat_all$file)] <- 10000
bm_dat_all$type[grepl('bgen', bm_dat_all$file)] <- 'bgen'
bm_dat_all$type[grepl('plink2', bm_dat_all$file)] <- 'plink2'
bm_dat_all$pgs_method[grepl('ptclump', bm_dat_all$file)] <- 'ptclump'
bm_dat_all$pgs_method[grepl('dbslmm', bm_dat_all$file)] <- 'dbslmm'
bm_dat_all$pgs_method[grepl('ldpred2', bm_dat_all$file)] <- 'ldpred2'
bm_dat_all$pgs_method[grepl('sbayesr', bm_dat_all$file)] <- 'sbayesr'
bm_dat_all$pgs_method[grepl('lassosum', bm_dat_all$file)] <- 'lassosum'
bm_dat_all$pgs_method[grepl('prscs', bm_dat_all$file)] <- 'prscs'
bm_dat_all$pgs_method[grepl('megaprs', bm_dat_all$file)] <- 'megaprs'
bm_dat_all$pgs_method[grepl('external', bm_dat_all$file)] <- 'external'
# Calculate mean time and memory for each rule
bm_dat_summary <- NULL
####
# Split by ncores
####
for (rule_i in c(
'prep_pgs_ptclump_i',
'prep_pgs_dbslmm_i',
'prep_pgs_prscs_i',
'prep_pgs_megaprs_i',
'prep_pgs_lassosum_i',
'prep_pgs_ldpred2_i',
'prep_pgs_sbayesr_i'
)) {
bm_dat_all_tmp <-
bm_dat_all[bm_dat_all$rule == rule_i, ]
for (ncores_i in unique(bm_dat_all_tmp$ncores)){
bm_dat_all_tmp_2 <-
bm_dat_all_tmp[bm_dat_all_tmp$ncores == ncores_i, ]
tmp <- data.table(
ncores = ncores_i,
rule = rule_i,
type = 'any',
n = 'any',
pgs_method = 'any',
mean_time = mean(bm_dat_all_tmp_2$s),
max_time = max(bm_dat_all_tmp_2$s),
mean_mem = mean(bm_dat_all_tmp_2$max_rss),
max_mem = max(bm_dat_all_tmp_2$max_rss)
)
bm_dat_summary <- rbind(bm_dat_summary, tmp)
}
}
####
# Split by ncores and n
####
# Sum across populations for each type
for (rule_i in c(
'target_pgs_i'
)) {
bm_dat_all_tmp <-
bm_dat_all[bm_dat_all$rule == rule_i, ]
for (n_i in unique(bm_dat_all_tmp$n)) {
for (ncores_i in unique(bm_dat_all_tmp$ncores)){
bm_dat_all_tmp_2 <-
bm_dat_all_tmp[bm_dat_all_tmp$ncores == ncores_i & bm_dat_all_tmp$n == n_i, ]
for (type_i in unique(bm_dat_all_tmp$type)) {
bm_dat_all_tmp_2$s[bm_dat_all_tmp_2$type == type_i]<-sum(bm_dat_all_tmp_2$s[bm_dat_all_tmp_2$type == type_i])
}
tmp <- data.table(
ncores = ncores_i,
rule = rule_i,
type = 'any',
n = n_i,
pgs_method = 'all',
mean_time = mean(bm_dat_all_tmp_2$s),
max_time = max(bm_dat_all_tmp_2$s),
mean_mem = mean(bm_dat_all_tmp_2$max_rss),
max_mem = max(bm_dat_all_tmp_2$max_rss)
)
bm_dat_summary <- rbind(bm_dat_summary, tmp)
}
}
}
####
# no split
####
for (rule_i in c(
'ref_pca_i',
'sumstat_prep_i',
'download_pgs_external',
'prep_pgs_external_i',
'score_reporter.txt',
'indiv_report_i'
)) {
bm_dat_all_tmp <-
bm_dat_all[bm_dat_all$rule == rule_i, ]
tmp <- data.table(
ncores = 'any',
rule = rule_i,
type = 'any',
n = 'any',
pgs_method = 'any',
mean_time = mean(bm_dat_all_tmp$s),
max_time = max(bm_dat_all_tmp$s),
mean_mem = mean(bm_dat_all_tmp$max_rss),
max_mem = max(bm_dat_all_tmp$max_rss)
)
bm_dat_summary <- rbind(bm_dat_summary, tmp)
}
####
# split by n and type
####
rule_i <- 'format_target_i'
bm_dat_all_tmp <-
bm_dat_all[bm_dat_all$rule == rule_i, ]
for (n_i in unique(bm_dat_all_tmp$n)) {
for (type_i in unique(bm_dat_all_tmp$type)) {
bm_dat_all_tmp2 <-
bm_dat_all_tmp[
bm_dat_all_tmp$n == n_i &
bm_dat_all_tmp$type == type_i, ]
tmp <- data.table(
ncores = 'any',
rule = rule_i,
type = type_i,
n = n_i,
pgs_method = 'any',
mean_time = mean(bm_dat_all_tmp2$s),
max_time = max(bm_dat_all_tmp2$s),
mean_mem = mean(bm_dat_all_tmp2$max_rss),
max_mem = max(bm_dat_all_tmp2$max_rss)
)
bm_dat_summary <- rbind(bm_dat_summary, tmp)
}
}
####
# split by n
####
for (rule_i in c('ancestry_inference_i','ancestry_reporter','pc_projection_i','sample_report_i','outlier_detection_i')) {
bm_dat_all_tmp <-
bm_dat_all[bm_dat_all$rule == rule_i, ]
for (n_i in unique(bm_dat_all_tmp$n)) {
bm_dat_all_tmp2 <-
bm_dat_all_tmp[
bm_dat_all_tmp$n == n_i, ]
tmp <- data.table(
ncores = 'any',
rule = rule_i,
type = 'any',
n = n_i,
pgs_method = 'any',
mean_time = mean(bm_dat_all_tmp2$s),
max_time = max(bm_dat_all_tmp2$s),
mean_mem = mean(bm_dat_all_tmp2$max_rss),
max_mem = max(bm_dat_all_tmp2$max_rss)
)
bm_dat_summary <- rbind(bm_dat_summary, tmp)
}
}
#################
# Plot
#################
####
# split by ncores
####
bm_dat_summary_i <- bm_dat_summary[bm_dat_summary$rule %in% c(
'prep_pgs_ptclump_i',
'prep_pgs_dbslmm_i',
'prep_pgs_prscs_i',
'prep_pgs_megaprs_i',
'prep_pgs_lassosum_i',
'prep_pgs_ldpred2_i',
'prep_pgs_sbayesr_i'
),]
bm_dat_summary_i$method <- gsub('_i', '', gsub('prep_pgs_','',bm_dat_summary_i$rule))
bm_dat_summary_i<-merge(bm_dat_summary_i, pgs_method_labels, by='method')
pgs_method_time <-
ggplot(bm_dat_summary_i, aes(x = label, y = mean_time, fill = ncores)) +
geom_bar(stat = "identity", position = "dodge") +
labs(x = "PGS Method", y = "Time (seconds)", fill='Cores') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
pgs_method_mem <-
ggplot(bm_dat_summary_i, aes(x = label, y = max_mem, fill = ncores)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "PGS Method", y = "Max Memory (Mb)", fill='Cores') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
png('Images/Benchmark/pgs_methods.png', res = 300, width = 1800, height = 1800, units = 'px')
plot_grid(pgs_method_time, pgs_method_mem, nrow=2)
dev.off()
####
# split by ncores and n
####
bm_dat_summary_i <- bm_dat_summary[bm_dat_summary$rule %in% c(
'target_pgs_i'
),]
target_pgs_time <-
ggplot(bm_dat_summary_i, aes(x = n, y = mean_time, fill = ncores)) +
geom_bar(stat = "identity", position = "dodge") +
labs(x = "N", y = "Time (seconds)", fill='Cores') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
target_pgs_mem <-
ggplot(bm_dat_summary_i, aes(x = n, y = max_mem, fill = ncores)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "N", y = "Max Memory (Mb)", fill='Cores') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
png('Images/Benchmark/target_pgs.png', res = 300, width = 1800, height = 1800, units = 'px')
plot_grid(target_pgs_time, target_pgs_mem, nrow=2)
dev.off()
####
# no split
####
bm_dat_summary_i <- bm_dat_summary[bm_dat_summary$rule %in% c(
'download_pgs_external',
'sumstat_prep_i',
'prep_pgs_external_i',
'ref_pca_i',
'indiv_report_i'
),]
no_split_time <-
ggplot(bm_dat_summary_i, aes(x = rule, y = mean_time, fill = rule)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Time (seconds)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
no_split_mem <-
ggplot(bm_dat_summary_i, aes(x = rule, y = max_mem, fill = rule)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Max Memory (Mb)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
png('Images/Benchmark/no_split.png', res = 300, width = 1800, height = 2200, units = 'px')
plot_grid(no_split_time, no_split_mem, nrow=2)
dev.off()
####
# split by n and type
####
bm_dat_summary_i <- bm_dat_summary[bm_dat_summary$rule %in% c('format_target_i'),]
n_type_split_time <-
ggplot(bm_dat_summary_i, aes(x = n, y = mean_time, fill = type)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Time (seconds)", fill = 'Type') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
n_type_split_mem <-
ggplot(bm_dat_summary_i, aes(x = n, y = max_mem, fill = type)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Max Memory (Mb)", fill = 'Type') +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
png('Images/Benchmark/n_type_split.png', res = 300, width = 1800, height = 1800, units = 'px')
plot_grid(n_type_split_time, n_type_split_mem, nrow=2)
dev.off()
####
# split by n
####
bm_dat_summary_i <-
bm_dat_summary[bm_dat_summary$rule %in% c(
'ancestry_inference_i',
'pc_projection_i',
'sample_report_i',
'outlier_detection_i'
), ]
n_split_time <-
ggplot(bm_dat_summary_i, aes(x = rule, y = mean_time, fill = n)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Time (seconds)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
n_split_mem <-
ggplot(bm_dat_summary_i, aes(x = rule, y = max_mem, fill = n)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "Rule", y = "Max Memory (Mb)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
png('Images/Benchmark/n_split.png', res = 300, width = 1800, height = 2200, units = 'px')
plot_grid(n_split_time, n_split_mem, nrow=2)
dev.off()Results
The pipeline was run using chromosome 22 only.
Show PGS Methods
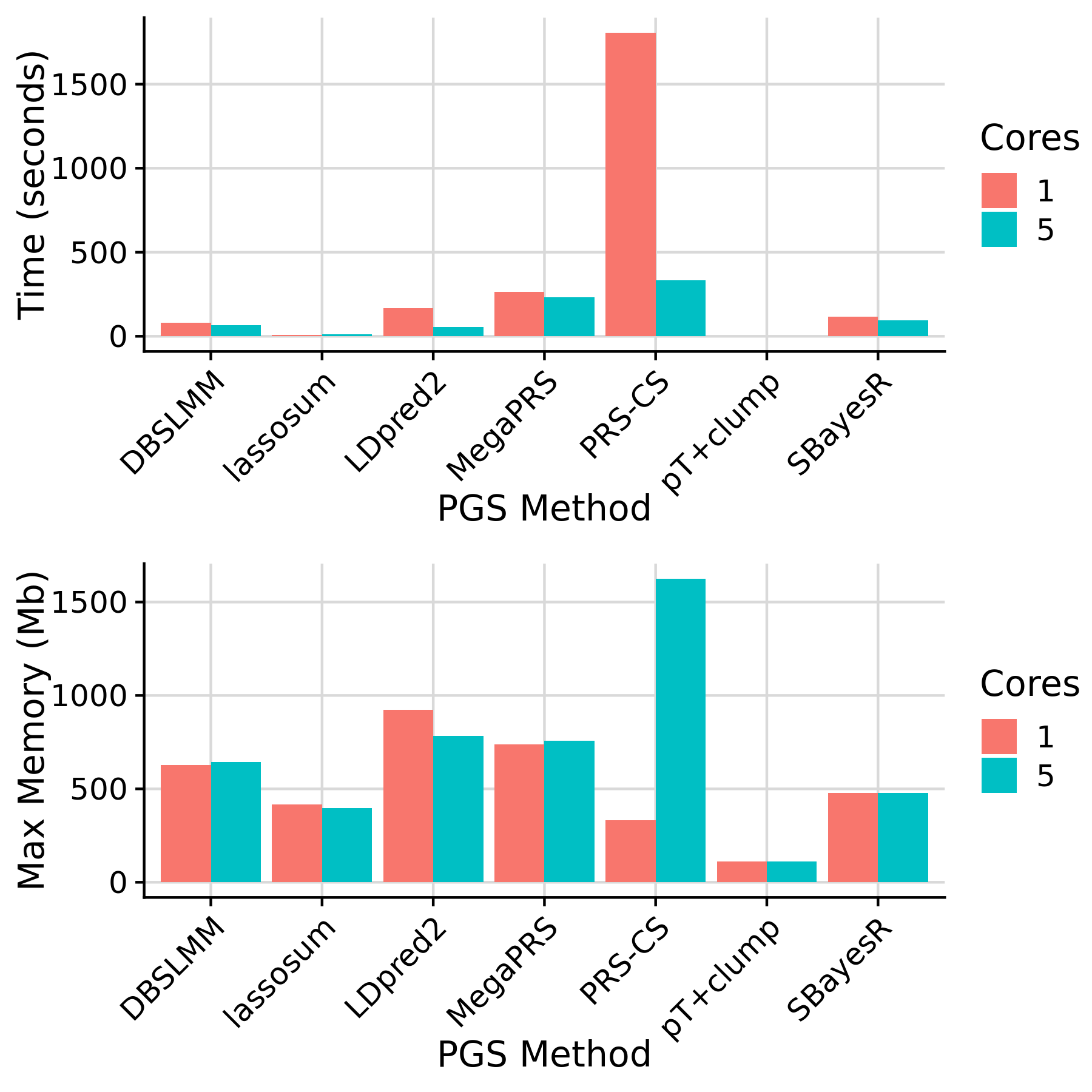
Rules affected by N cores and PGS method
Note: SBayesR and DBSLMM would capitalise on multiple cores when running on multiple chromosomes. pT+clump is not implemented in parallel, since they are already fast.
Show target scoring
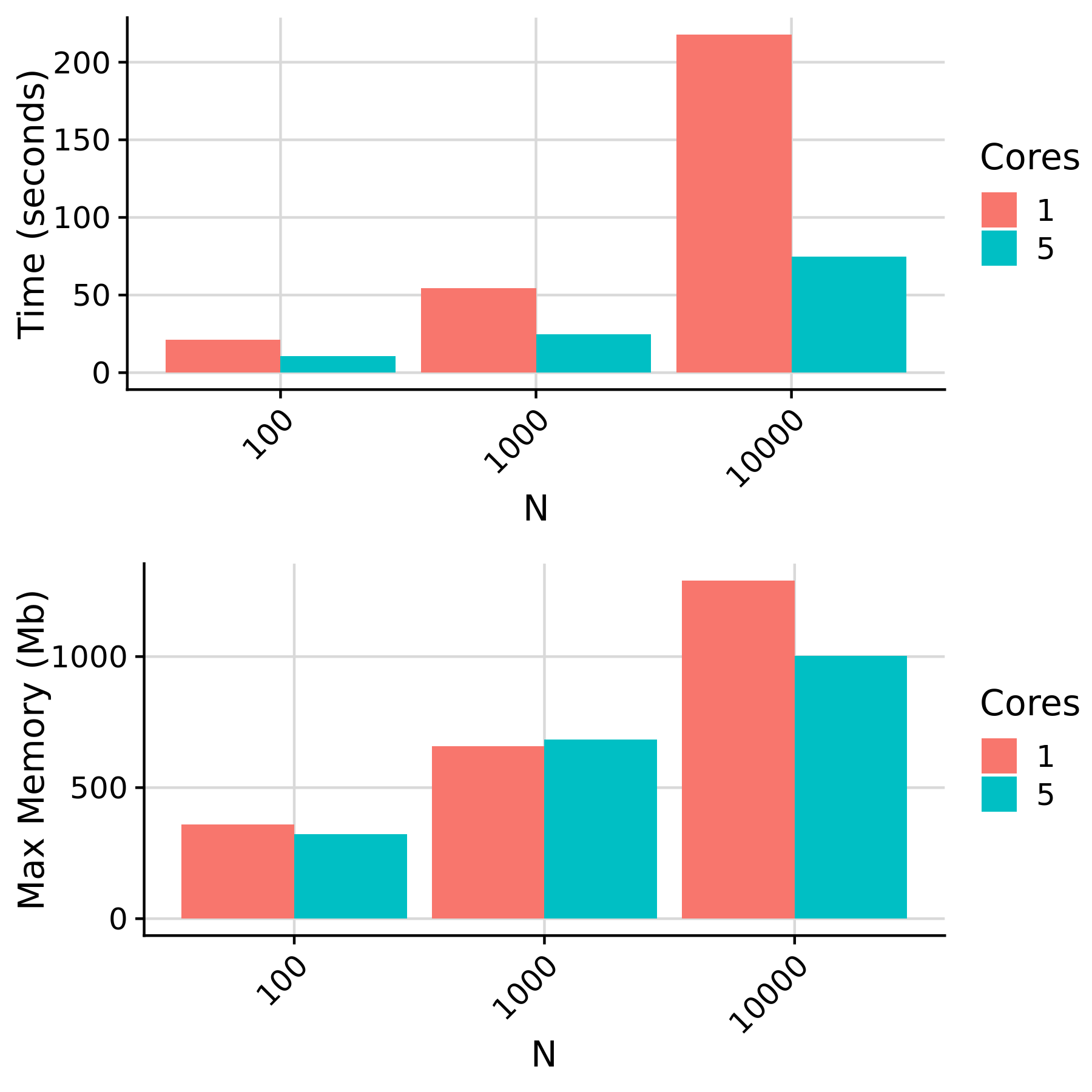
Rules affected by N cores and target sample size
Note: target_pgs_i resources vary according to the number of pgs_methods and scores requested. In this benchmark, 4586 scores were computed for each individual.
Show rules with constant time/memory
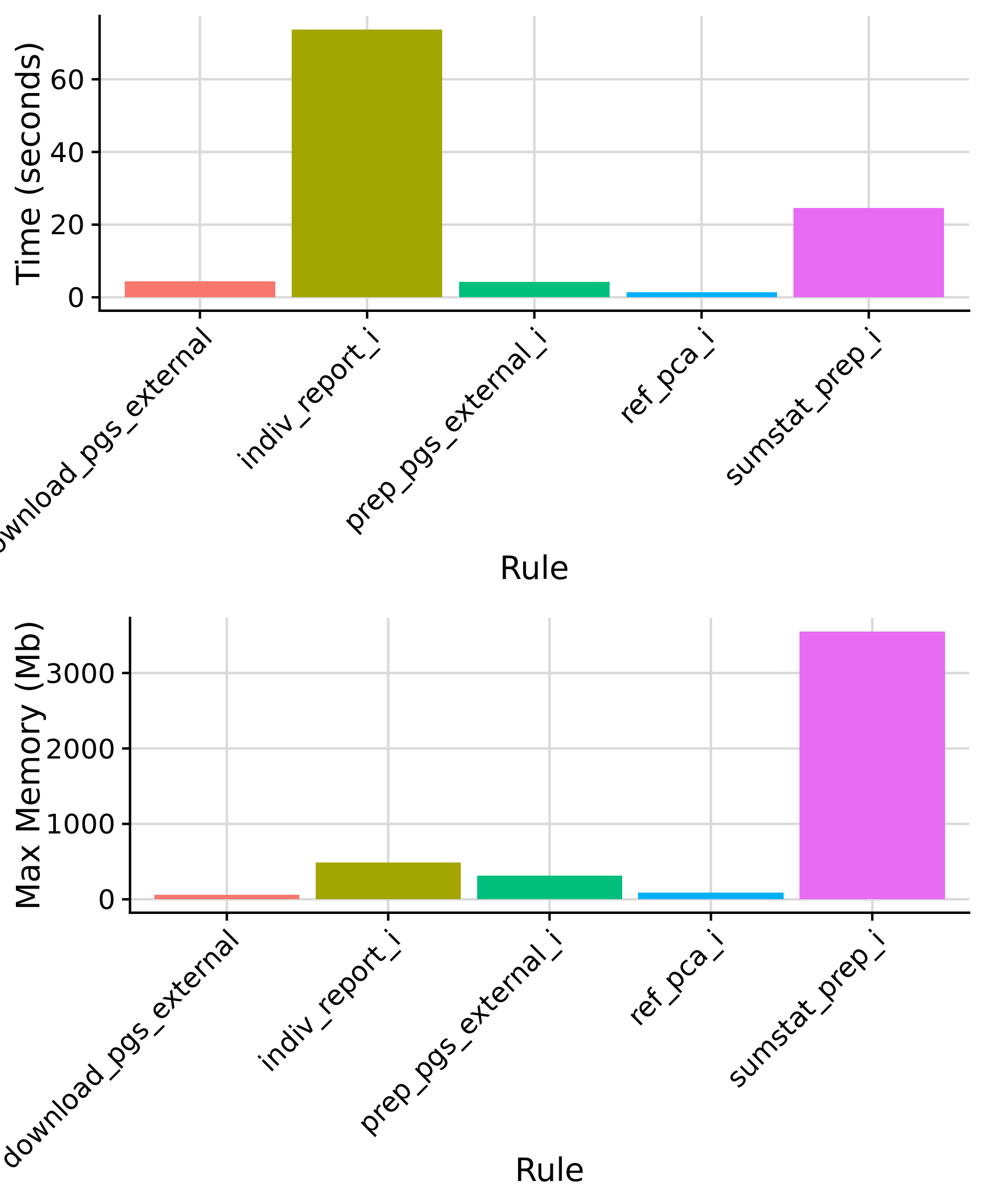
Rules with constant time/memory
Show rules affected by N
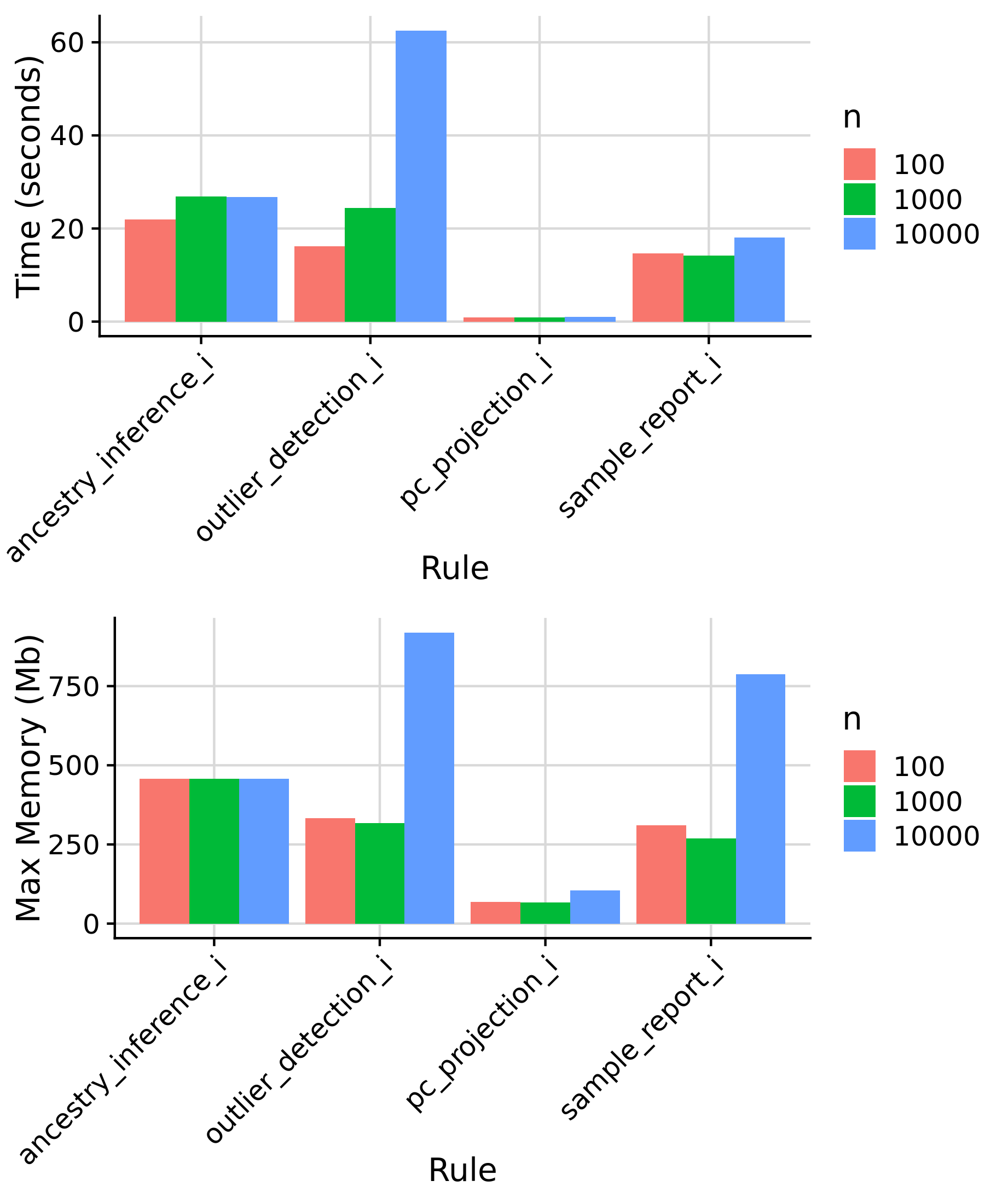
Rules affected by target sample size
Show rules affected by N and Type (format_target_i)
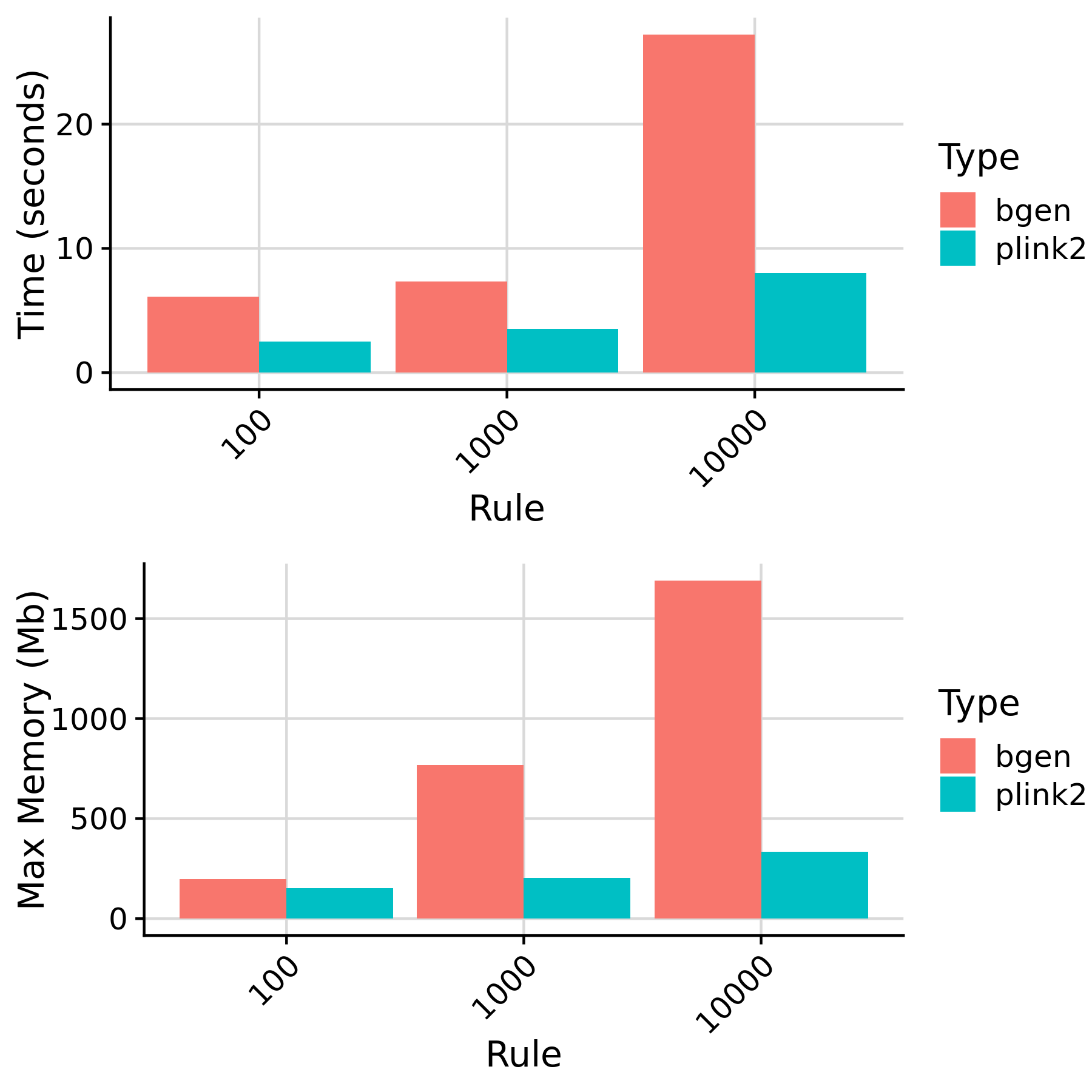
Rules affected by target sample size and genotype data format
Required storage
This shows the disk space required for the reference data:
38G gctb_ref
17M hm3_snplist
2.3G ldak_bld
1.0K ldak_highld
119M ldak_map
459K ld_blocks
7.2G ldpred2_ref
44M ldsc_ref
21M logs
4.4G prscs_ref
1.1G refIn total this is ~50G, most of which is the GCTB reference required for SBayesR.
Genome-wide benchmark
We also performed a benchmark of PGS methods genome-wide using European GWAS summary statistics from Yengo et al. This was carried out using default settings in GenoPred, with 10 cores allocated to each PGS method, except pT+clump and DBSLMM which are run on a single core.
Show PGS methods benchmark
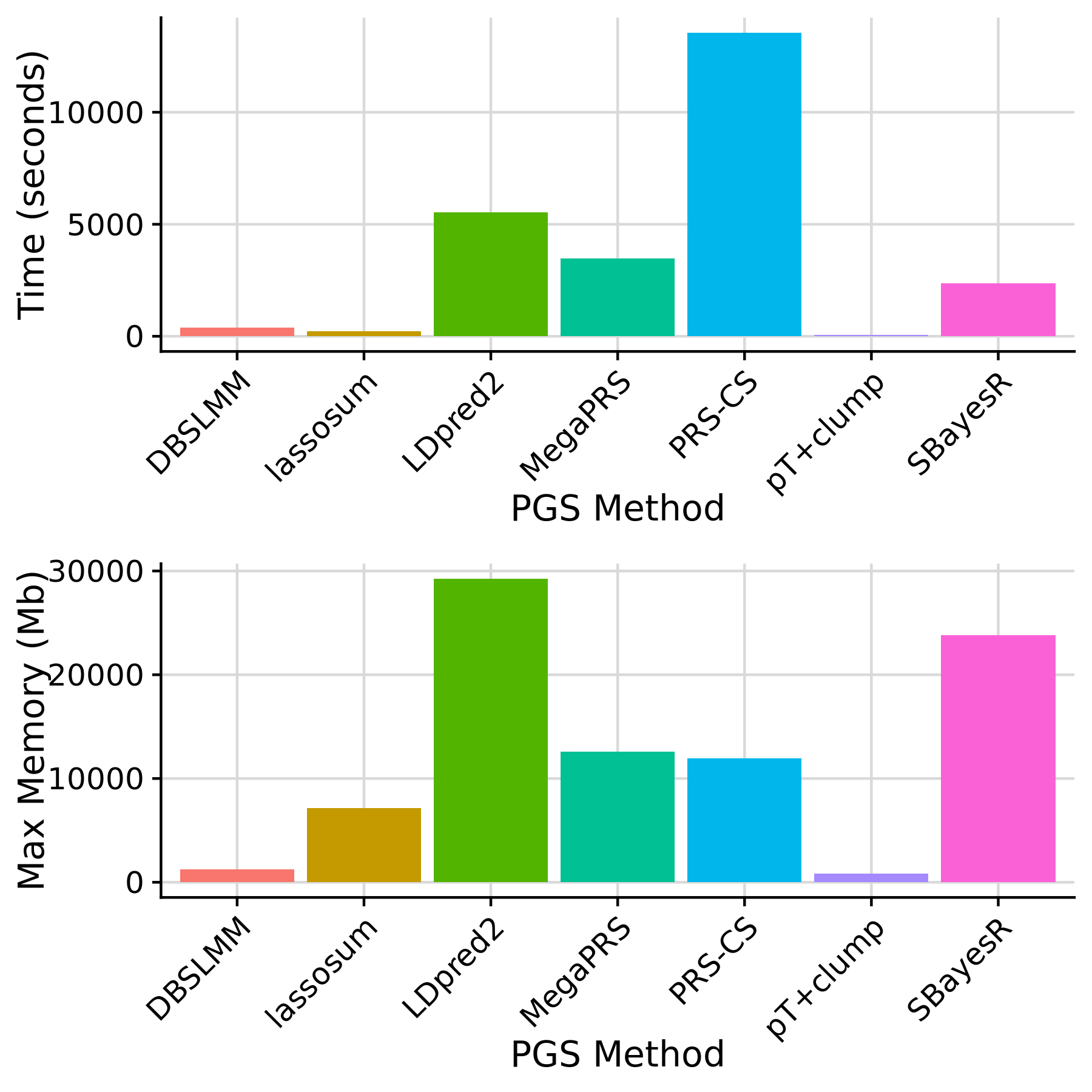
PGS methods run genome-wide
Note: pT+clump is run using 1 core. All other methods are run using 10 cores.