OpenSNP Benchmark
Overview
We used the publicly available OpenSNP dataset to test the GenoPred pipeline. We used GWAS sumstats for Height from the Yengo et al. GWAS. We used results derived using European and East Asian individuals. We ran the original version of GenoPred (V1) and the updated version of GenoPred (v2.0.0) to check results were consistent.
As can be seen in the figure below, the GenoPred returned polygenic scores performing as expected, and results were consistent across version of GenoPred.
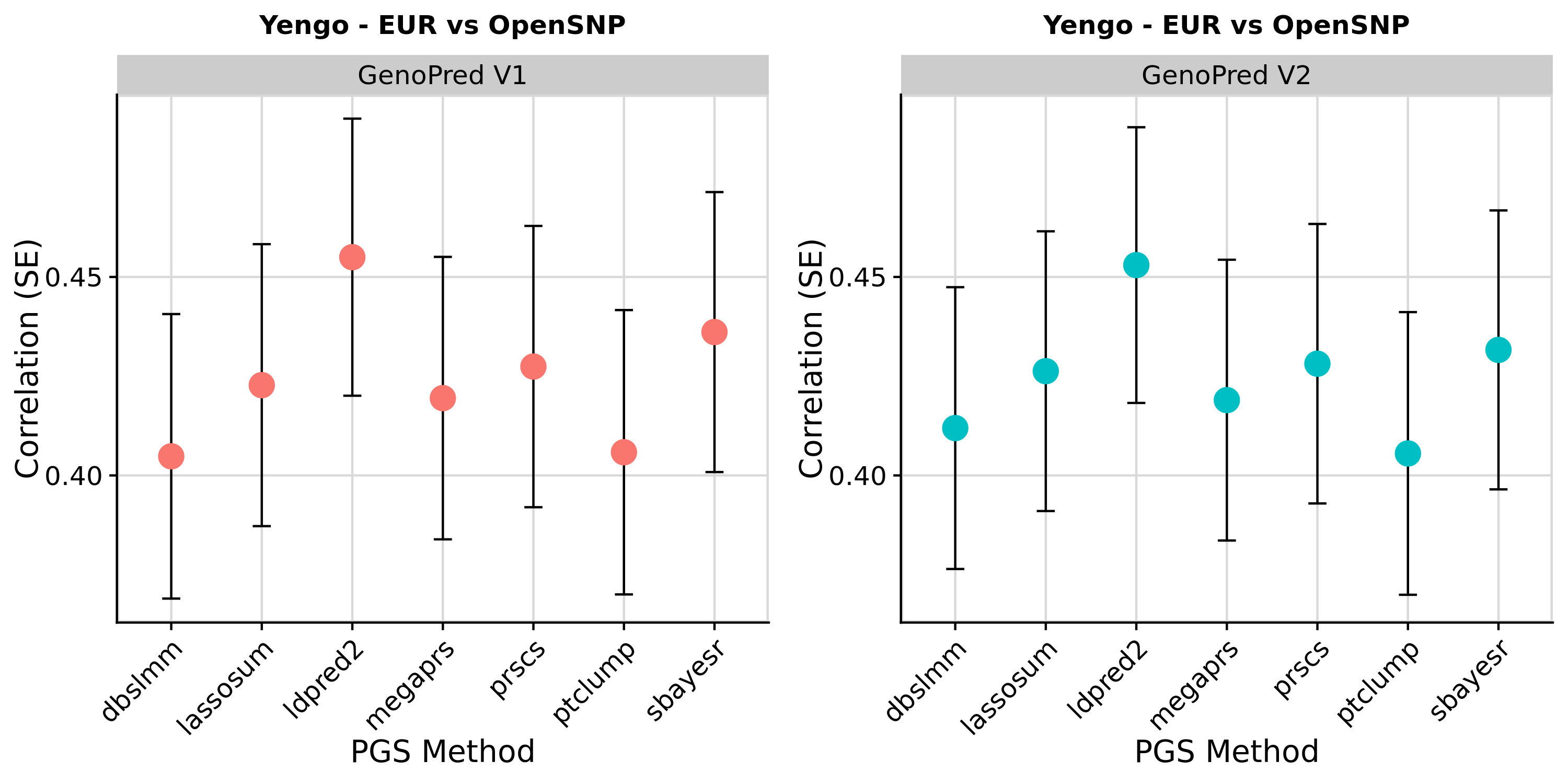
Performance of PGS methods using European Yengo GWAS in OpenSNP European individuals
GenoPred V2
Prepare input
Download genotypes
Show code
mkdir -p /users/k1806347/oliverpainfel/Data/OpenSNP/raw
cd /users/k1806347/oliverpainfel/Data/OpenSNP/raw
wget https://zenodo.org/records/1442755/files/CrowdAI_v3.tar.gz?download=1
mv 'CrowdAI_v3.tar.gz?download=1' CrowdAI_v3.tar.gz
tar -xvzf CrowdAI_v3.tar.gz
rm CrowdAI_v3.tar.gz
# Use both training and testing data to maximise sample size for testing.Prepare phenotype
Show code
library(data.table)
# Training
train_dat <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/training_set_details.txt')
train_dat$FID <- train_dat$id
train_dat$IID <- train_dat$id
train_dat <- train_dat[, c('FID', 'IID', 'height'), with = F]
# Test
test_dat <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/test_set_details_SECRET.txt')
test_dat$FID <- test_dat$id
test_dat$IID <- test_dat$id
test_dat <- test_dat[, c('FID', 'IID', 'height'), with = F]
all_dat<-rbind(train_dat, test_dat)
dir.create('/users/k1806347/oliverpainfel/Data/OpenSNP/processed/pheno', recursive = T)
write.table(
all_dat,
'/users/k1806347/oliverpainfel/Data/OpenSNP/processed/pheno/height.txt',
col.names = T,
row.names = F,
quote = F
)Split VCF by chromosome
Show code
module add bcftools/1.12-gcc-13.2.0-python-3.11.6
# Create index
bcftools index /users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_train.vcf.gz
bcftools index /users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_test.vcf.gz
# Merge the vcfs for training and testing data
bcftools merge \
/users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_train.vcf.gz \
/users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_test.vcf.gz \
-o /users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_merged.vcf.gz
# Now, split by chromosome using plink2
# Run on the command line within pipeline conda environment
mkdir /users/k1806347/oliverpainfel/Data/OpenSNP/processed/geno
for chr in $(seq 1 22);do
/users/k1806347/oliverpainfel/Software/plink2 \
--vcf /users/k1806347/oliverpainfel/Data/OpenSNP/raw/CrowdAI_v3/fullset/genotyping_data_fullset_merged.vcf.gz \
--chr ${chr} \
--out /users/k1806347/oliverpainfel/Data/OpenSNP/processed/geno/opensnp_merged.chr${chr} \
--export vcf bgz
doneDownload height GWAS
Show code
# These are from the Yengo 2022 paper
mkdir -p /users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test
wget --no-check-certificate -O /users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eur.txt https://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90245001-GCST90246000/GCST90245992/GCST90245992_buildGRCh37.tsv
wget --no-check-certificate -O /users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eas.txt https://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90245001-GCST90246000/GCST90245991/GCST90245991_buildGRCh37.tsvCreate gwas_list, target_list and config
Show code
setwd('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/pipeline')
library(data.table)
# Create config file
conf <- c(
'outdir: /users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred/test6',
'config_file: misc/opensnp/config.yaml',
'gwas_list: misc/opensnp/gwas_list.txt',
'score_list: misc/opensnp/score_list.txt',
'target_list: misc/opensnp/target_list.txt',
'gwas_groups: misc/opensnp/gwas_groups.txt',
"pgs_methods: ['ptclump','dbslmm','prscs','sbayesr','lassosum','ldpred2','megaprs','prscsx']",
'testing: NA'
)
dir.create('misc/opensnp/', recursive = T)
dir.create('/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred', recursive = T)
write.table(conf, 'misc/opensnp/config.yaml', col.names = F, row.names = F, quote = F)
# Create target_list
target_list <- fread('example_input/target_list.txt')
target_list <- rbind(target_list,
data.table(name = 'opensnp',
path = '/users/k1806347/oliverpainfel/Data/OpenSNP/processed/geno/opensnp_merged',
type = 'vcf',
indiv_report = F))
target_list <- target_list[target_list$name == 'opensnp', ]
write.table(target_list, 'misc/opensnp/target_list.txt', col.names = T, row.names = F, quote = F, sep = ' ')
# Create gwas_list
gwas_list <- fread('example_input/gwas_list.txt')
gwas_list<-rbind(gwas_list,
data.table(name='yengo_eur',
path = '/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eur.txt',
population = 'EUR',
n = NA,
sampling = NA,
prevalence = NA,
mean = NA,
sd = NA,
label = "\"Yengo 2022 Height EUR\""))
gwas_list <- rbind(gwas_list,
data.table(name = 'yengo_eas',
path = '/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eas.txt',
population = 'EAS',
n = NA,
sampling = NA,
prevalence = NA,
mean = NA,
sd = NA,
label = "\"Yengo 2022 Height EAS\""))
gwas_list <- gwas_list[gwas_list$name %in% c('yengo_eur', 'yengo_eas'), ]
write.table(gwas_list, 'misc/opensnp/gwas_list.txt', col.names = T, row.names = F, quote = F, sep = ' ')
# Create gwas_groups
gwas_groups <- data.frame(
name = 'height',
gwas = 'yengo_eur,yengo_eas',
label = "\"Yengo 2022 Height EUR+EAS\""
)
write.table(gwas_groups, 'misc/opensnp/gwas_groups.txt', col.names = T, row.names = F, quote = F, sep = ' ')
# Create score_list
score_list <- data.frame(
name = 'PGS002804',
path = NA,
label = "\"Yengo 2022 Height EUR PGSC\""
)
write.table(score_list, 'misc/opensnp/score_list.txt', col.names=T, row.names=F, quote=F, sep=' ')Run GenoPred
Show code
snakemake --profile slurm --use-conda --configfile=misc/opensnp/config.yaml output_all -n
snakemake --profile slurm --use-conda --configfile=misc/opensnp/config.yaml outlier_detection -n Check time and memory requirements
Show code
library(data.table)
library(ggplot2)
library(cowplot)
# Read in configuration specific benchmark files
bm_files_i <-
paste0(
'/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred/test6/reference/benchmarks/',
list.files('/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred/test6/reference/benchmarks/')
)
# Read in benchmark files
bm_dat_all <- do.call(rbind, lapply(bm_files_i, function(file) {
tmp <- fread(file)
tmp$file <- basename(file)
return(tmp)
}))
# Create rule column
bm_dat_all$rule <- gsub('-.*','',bm_dat_all$file)
#####
# PGS methods
#####
# Look at the memory required for each PGS method using the EUR GWAS
bm_dat_pgs_prep <-
bm_dat_all[grepl('prep_pgs', bm_dat_all$file), ]
bm_dat_yengo_eur$method <- gsub('_i', '', gsub('prep_pgs_','',bm_dat_yengo_eur$rule))
bm_dat_yengo_eur<-merge(bm_dat_yengo_eur, pgs_method_labels, by='method')
pgs_method_time <-
ggplot(bm_dat_yengo_eur, aes(x = label, y = s, fill = label)) +
geom_bar(stat = "identity", position = "dodge") +
labs(x = "PGS Method", y = "Time (seconds)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
pgs_method_mem <-
ggplot(bm_dat_yengo_eur, aes(x = label, y = max_rss, fill = label)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "PGS Method", y = "Max Memory (Mb)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
png('Images/OpenSNP/time_cpu_bench_pgs_methods.png', res = 300, width = 1800, height = 1800, units = 'px')
plot_grid(pgs_method_time, pgs_method_mem, nrow=2)
dev.off()
write.csv(bm_dat_yengo_eur, 'Images/OpenSNP/time_cpu_bench_pgs_methods.csv', row.names=F, quote=F)
# Look at time and memory for PRS-CSx analysis
bm_dat_height <-
bm_dat_all[grepl('prep_pgs', bm_dat_all$file) &
grepl('height.txt', bm_dat_all$file), ]
bm_dat_height$method <- gsub('_i', '', gsub('prep_pgs_','',bm_dat_height$rule))
bm_dat_height<-merge(bm_dat_height, pgs_method_labels, by='method')
pgs_method_time <-
ggplot(bm_dat_height, aes(x = label, y = s, fill = label)) +
geom_bar(stat = "identity", position = "dodge") +
labs(x = "PGS Method", y = "Time (seconds)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
pgs_method_mem <-
ggplot(bm_dat_height, aes(x = label, y = max_rss, fill = label)) +
geom_bar(stat = "identity", position="dodge") +
labs(x = "PGS Method", y = "Max Memory (Mb)") +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position="none")
png('Images/OpenSNP/time_cpu_bench_pgs_methods_prscsx.png', res = 300, width = 800, height = 1800, units = 'px')
plot_grid(pgs_method_time, pgs_method_mem, nrow=2)
dev.off()
write.csv(bm_dat_height, 'Images/OpenSNP/time_cpu_bench_pgs_methods_prscsx.csv', row.names=F, quote=F)Show PGS methods benchmark
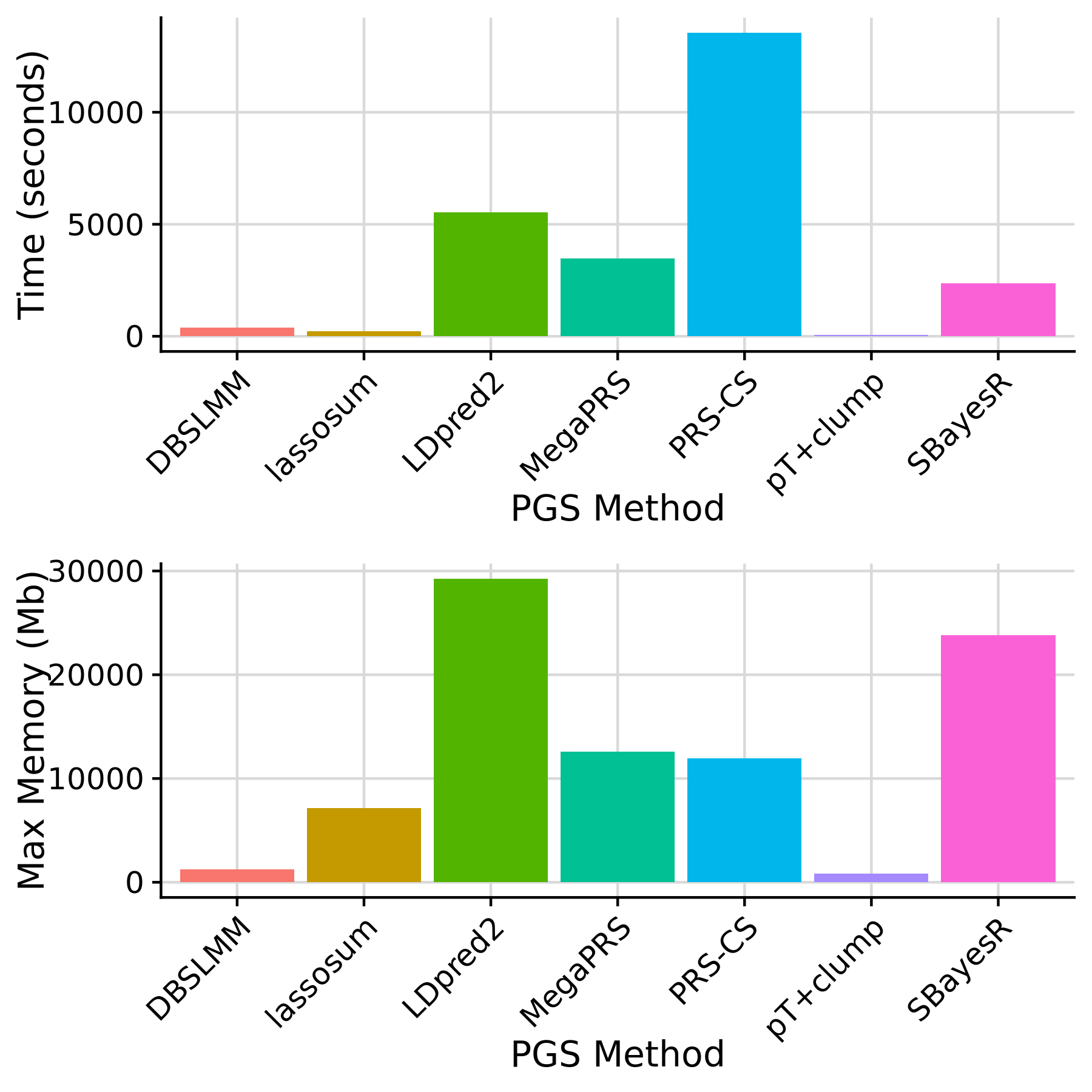
Time and max. memory used by each PGS method
Note: pT+clump is run using 1 core. All other methods are run using 10 cores.
Show PRS-CSx benchmark
Time and max. memory used by PRS-CSx
Evaluate PGS
Show code
# Test correlation between PGS and observed height
setwd('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/pipeline/')
library(data.table)
library(ggplot2)
library(cowplot)
source('../functions/misc.R')
source_all('../functions')
# Read in pheno data
pheno <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/processed/pheno/height.txt')
# Read in PGS
pgs <- read_pgs(config = 'misc/opensnp/config.yaml', name = 'opensnp')$opensnp
# Estimate correlation between pheno and pgs
cor <- NULL
for(pop_i in names(pgs)){
for(gwas_i in names(pgs[[pop_i]])){
for(pgs_method_i in names(pgs[[pop_i]][[gwas_i]])){
pgs_i <- pgs[[pop_i]][[gwas_i]][[pgs_method_i]]
pheno_pgs<-merge(pheno, pgs_i, by = c('FID','IID'))
for(model_i in names(pgs_i)[-1:-2]){
y <- scale(pheno_pgs$height)
x <- scale(pheno_pgs[[model_i]])
if(all(is.na(x))){
next
}
coef_i <- coef(summary(mod <- lm(y ~ x)))
tmp <- data.table(
pop = pop_i,
gwas = gwas_i,
pgs_method = pgs_method_i,
name = model_i,
r = coef_i[2,1],
se = coef_i[2,2],
p = coef_i[2,4],
n = nobs(mod))
cor <- rbind(cor, tmp)
}
}
}
}
# Save the results
dir.create('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc')
write.csv(
cor,
'/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-yengo-assoc.csv',
row.names = F
)
# The European sample is the only one large enough for interpretable results. All other populations are <20 individuals
# Subset EUR results
cor_eur <- cor[cor$pop == 'EUR', ]
# Restrict to best and and pseudoval only
cor_eur_subset <- NULL
for(pop_i in unique(cor_eur$pop)){
for(gwas_i in unique(cor_eur$gwas[cor_eur$pop == pop_i])){
for(pgs_method_i in unique(cor_eur$pgs_method[cor_eur$pop == pop_i & cor_eur$gwas == gwas_i])){
# Subset relevant results
cor_eur_i <- cor_eur[
cor_eur$pop == pop_i &
cor_eur$gwas == gwas_i &
cor_eur$pgs_method == pgs_method_i,]
# Top R
if(pgs_method_i %in% c('ptclump','ldpred2','megaprs','prscs','lassosum','dbslmm')){
top_i <- cor_eur_i[which(cor_eur_i$r == max(cor_eur_i$r, na.rm = T))[1],]
top_i$model <- 'Top'
cor_eur_subset <- rbind(cor_eur_subset, top_i)
}
if(pgs_method_i %in% c('prscsx')){
for(targ_i in unique(gsub('.*_','', gsub('_phi.*','',cor_eur_i$name)))){
cor_eur_k<-cor_eur_i[grepl(paste0(targ_i,'_phi_'), cor_eur_i$name),]
top_i <- cor_eur_k[which(cor_eur_k$r == max(cor_eur_k$r, na.rm = T))[1],]
top_i$model <- 'Top'
top_i$pgs_method<-paste0(top_i$pgs_method, " (", targ_i, ")")
cor_eur_subset <- rbind(cor_eur_subset, top_i)
}
}
# PseudoVal
if(pgs_method_i %in% c('ptclump','sbayesr','ldpred2','megaprs','prscs','lassosum','dbslmm','prscsx')){
pseudo_param <- find_pseudo(config = 'misc/opensnp/config.yaml', gwas = gwas_i, pgs_method = pgs_method_i)
pseudo_i <- cor_eur_i[grepl(paste0(pseudo_param,'$'), cor_eur_i$name),]
pseudo_i$model <- 'Pseudo'
if(pgs_method_i %in% c('prscsx')){
pseudo_i$pgs_method<-paste0(pseudo_i$pgs_method, " (", gsub('.*_','', gsub(paste0('_',pseudo_param), '', pseudo_i$name)), ")")
}
cor_eur_subset <- rbind(cor_eur_subset, pseudo_i)
}
# External
external_tmp<-cor_eur_i[cor_eur_i$pgs_method == 'external',]
external_tmp$model <- 'External'
cor_eur_subset <- rbind(cor_eur_subset, external_tmp)
}
}
}
# Plot the results
cor_eur_subset$model <- factor(cor_eur_subset$model, levels = c('Top','Pseudo','External'))
dir.create('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP')
# yengo_eur
plot_obj_eur <-
ggplot(cor_eur_subset[cor_eur_subset$gwas == 'yengo_eur',], aes(x = pgs_method, y = r, fill = model)) +
geom_bar(stat = "identity", position = position_dodge2(preserve = "single"), width = 0.7) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
fill = 'Model',
title = paste0("Yengo - EUR vs OpenSNP - EUR\n(N = ", cor_eur_subset$n[1], ")")
) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12))
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-yengo_eur.png',
units = 'px',
width = 2000,
height = 1000,
res = 300)
plot_obj_eur
dev.off()
# yengo_eas
plot_obj_eas <-
ggplot(cor_eur_subset[cor_eur_subset$gwas == 'yengo_eas',], aes(x = pgs_method, y = r, fill = model)) +
geom_bar(stat = "identity", position = position_dodge2(preserve = "single"), width = 0.7) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
fill = 'Model',
title = paste0("Yengo - EAS vs OpenSNP - EUR\n(N = ", cor_eur_subset$n[1], ")")
) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12))
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-yengo_eas.png',
units = 'px',
width = 1200,
height = 1000,
res = 300)
plot_obj_eas
dev.off()
# Make a plot comparing the yengo_eur results to the score file from PGS-catalogue
not_external <- unique(cor_eur_subset$pgs_method[cor_eur_subset$pgs_method != 'external'])
cor_eur_subset$pgs_method <- factor(cor_eur_subset$pgs_method, levels=c(not_external,'external'))
plot_obj_ext <-
ggplot(cor_eur_subset[cor_eur_subset$gwas == 'yengo_eur' | cor_eur_subset$gwas == 'PGS002804',], aes(x = pgs_method, y = r, fill = model)) +
geom_bar(stat = "identity", position = position_dodge2(preserve = "single"), width = 0.7) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
fill = 'Model',
title = paste0("Yengo - EUR vs OpenSNP - EUR\n(N = ", cor_eur_subset$n[1], ")")
) +
theme_half_open() +
background_grid() +
geom_vline(xintercept = 7.5, linetype = 'dashed') +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12))
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-yengo_eur-external.png',
units = 'px',
width = 2000,
height = 1000,
res = 300)
plot_obj_ext
dev.off()
# Subset EAS results
cor_eas <- cor[cor$pop == 'EAS', ]
# Restrict to best and and pseudoval only
cor_eas_subset <- NULL
for(pop_i in unique(cor_eas$pop)){
for(gwas_i in unique(cor_eas$gwas[cor_eas$pop == pop_i])){
for(pgs_method_i in unique(cor_eas$pgs_method[cor_eas$pop == pop_i & cor_eas$gwas == gwas_i])){
# Subset relevant results
cor_eas_i <- cor_eas[
cor_eas$pop == pop_i &
cor_eas$gwas == gwas_i &
cor_eas$pgs_method == pgs_method_i,]
# Top R
if(pgs_method_i %in% c('ptclump','ldpred2','megaprs','prscs','lassosum','dbslmm')){
top_i <- cor_eas_i[which(cor_eas_i$r == max(cor_eas_i$r, na.rm = T))[1],]
top_i$model <- 'Top'
cor_eas_subset <- rbind(cor_eas_subset, top_i)
}
if(pgs_method_i %in% c('prscsx')){
for(targ_i in unique(gsub('.*_','', gsub('_phi.*','',cor_eas_i$name)))){
cor_eas_k<-cor_eas_i[grepl(paste0(targ_i,'_phi_'), cor_eas_i$name),]
top_i <- cor_eas_k[which(cor_eas_k$r == max(cor_eas_k$r, na.rm = T))[1],]
top_i$model <- 'Top'
top_i$pgs_method<-paste0(top_i$pgs_method, " (", targ_i, ")")
cor_eas_subset <- rbind(cor_eas_subset, top_i)
}
}
# PseudoVal
if(pgs_method_i %in% c('ptclump','sbayesr','ldpred2','megaprs','prscs','lassosum','dbslmm','prscsx')){
pseudo_param <- find_pseudo(config = 'misc/opensnp/config.yaml', gwas = gwas_i, pgs_method = pgs_method_i)
pseudo_i <- cor_eas_i[grepl(paste0(pseudo_param,'$'), cor_eas_i$name),]
pseudo_i$model <- 'Pseudo'
if(pgs_method_i %in% c('prscsx')){
pseudo_i$pgs_method<-paste0(pseudo_i$pgs_method, " (", gsub('.*_','', gsub(paste0('_',pseudo_param), '', pseudo_i$name)), ")")
}
cor_eas_subset <- rbind(cor_eas_subset, pseudo_i)
}
# External
external_tmp<-cor_eas_i[cor_eas_i$pgs_method == 'external',]
external_tmp$model <- 'External'
cor_eas_subset <- rbind(cor_eas_subset, external_tmp)
}
}
}
# Make a plot including PRS-CSx comparing in EUR and EAS OpenSNP individuals
cor_eur_eas_subset<-rbind(cor_eur_subset, cor_eas_subset)
cor_eur_eas_subset$pop<-paste0(cor_eur_eas_subset$pop, " (N = ", cor_eur_eas_subset$n, ")")
plot_obj_prscsx <-
ggplot(cor_eur_eas_subset[cor_eur_eas_subset$gwas %in% c('yengo_eur','yengo_eas','height'),], aes(x = pgs_method, y = r, colour = model)) +
geom_point(stat = "identity", size = 4, position = position_dodge(width = 0.7)) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
colour = 'Model',
) +
theme_half_open() +
panel_border() +
background_grid() +
facet_grid(pop ~ gwas, scales = 'free', space = 'free_x') +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12))
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-prscsx.png',
units = 'px',
width = 3000,
height = 1500,
res = 300)
plot_obj_prscsx
dev.off()Show results in OpenSNP
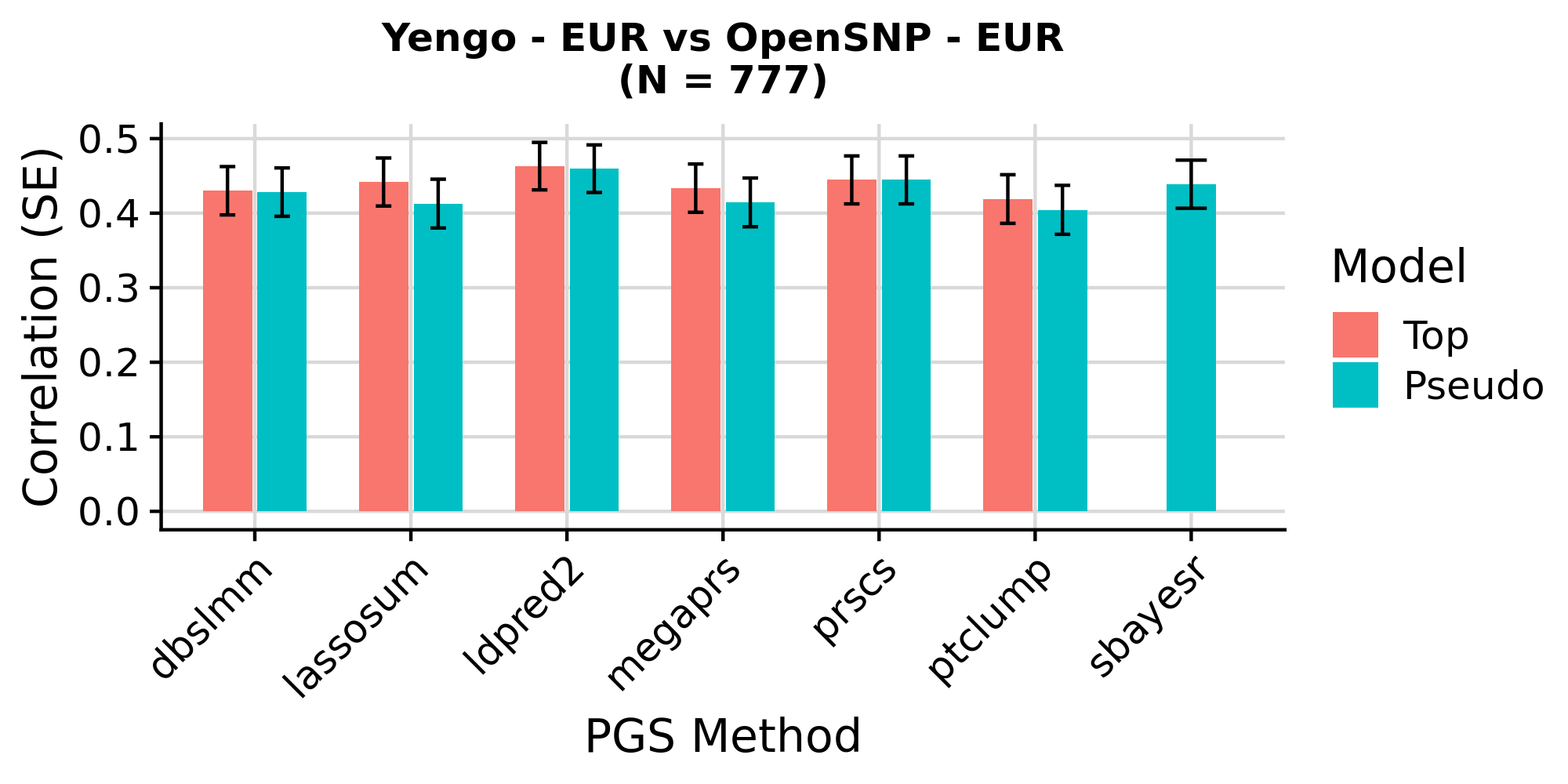
Yengo - EUR PGS correlation with height in EUR OpenSNP
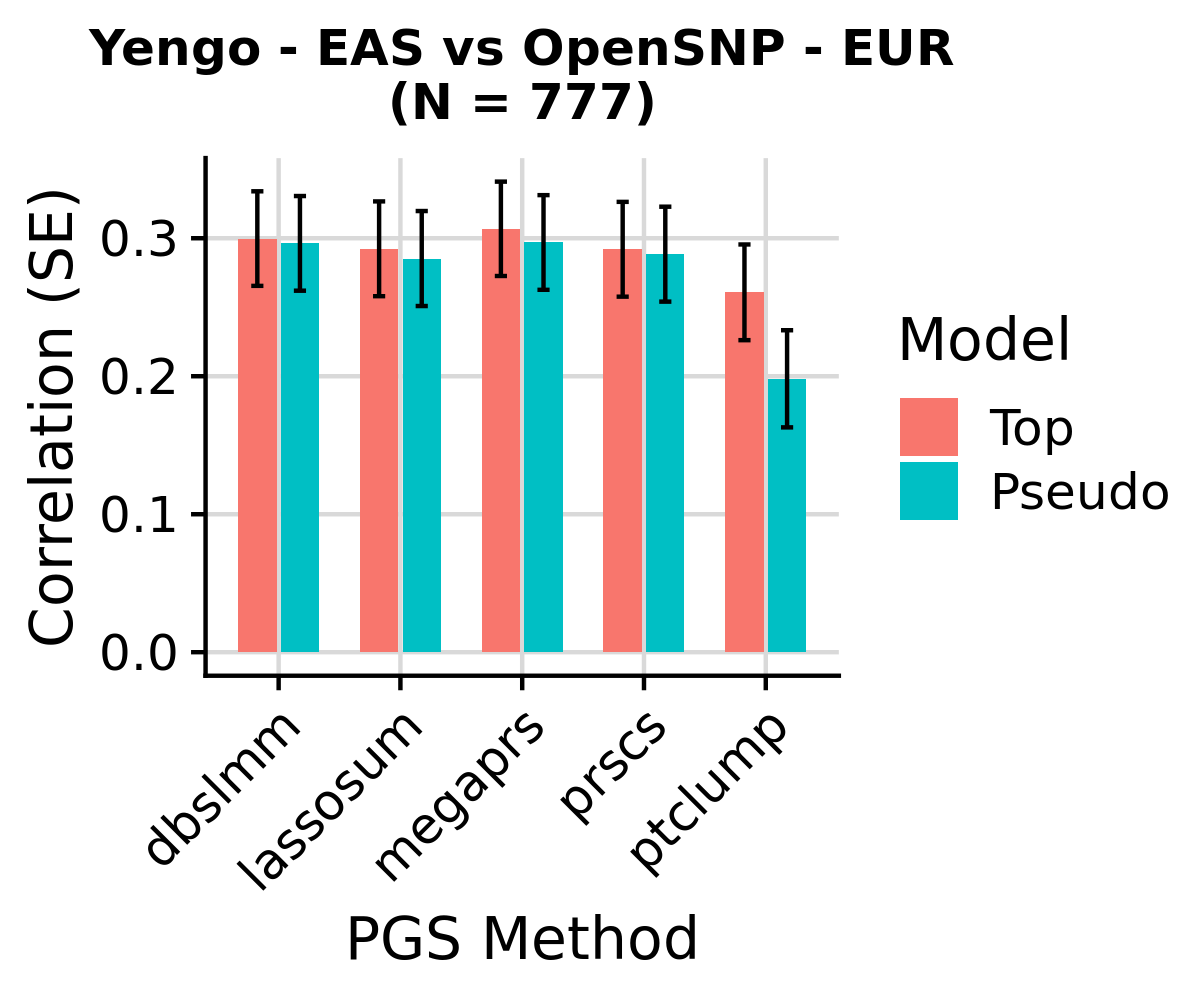
Yengo - EAS PGS correlation with height in EUR OpenSNP
Show results in OpenSNP including score from PGS catalogue
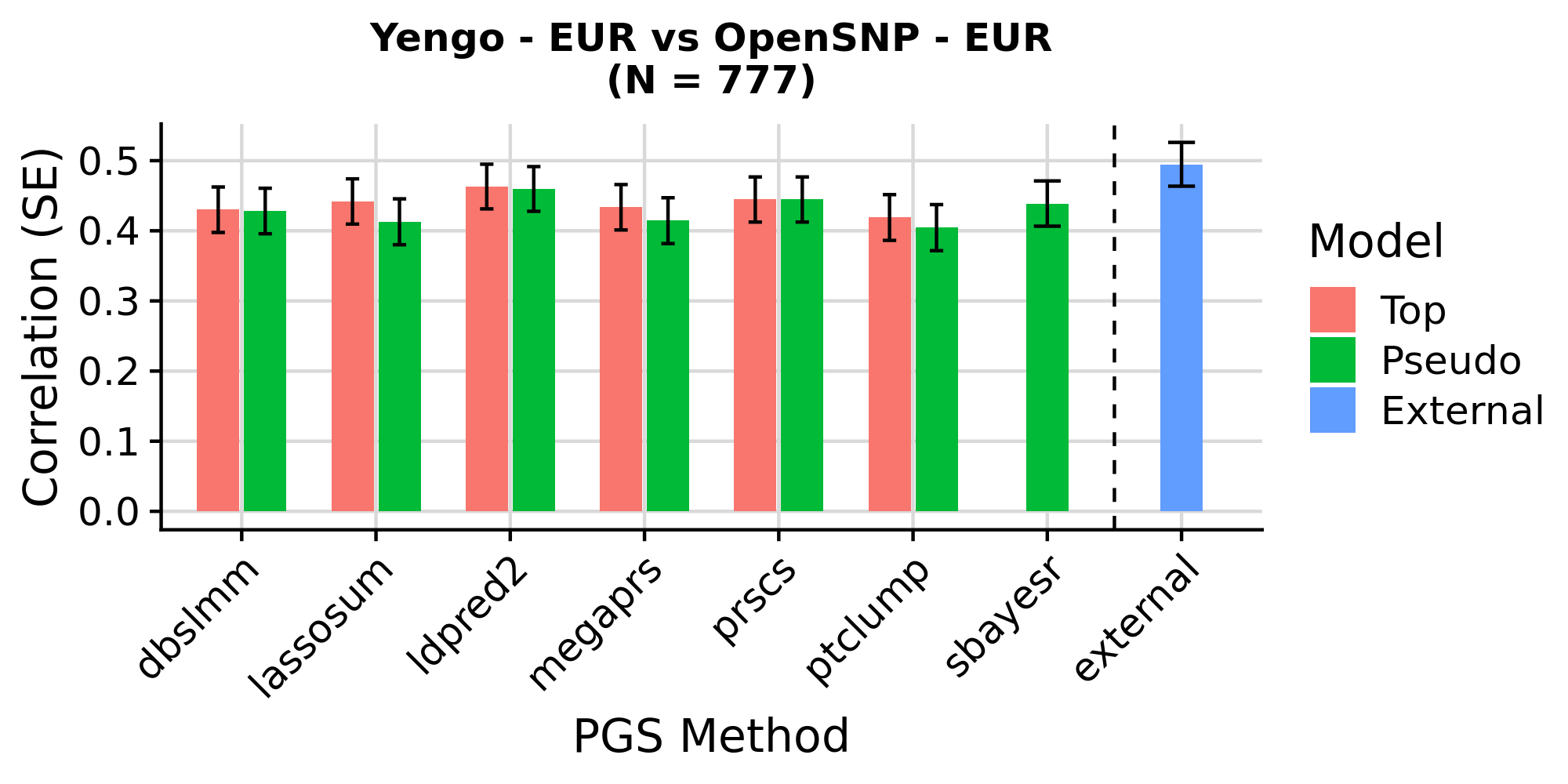
Yengo - EUR PGS correlation with height in EUR OpenSNP - Includes externally derived PGS
Note. ‘external’ is based on a larger GWAS (incl. 23andMe)
Show results in OpenSNP when using PRS-CSx
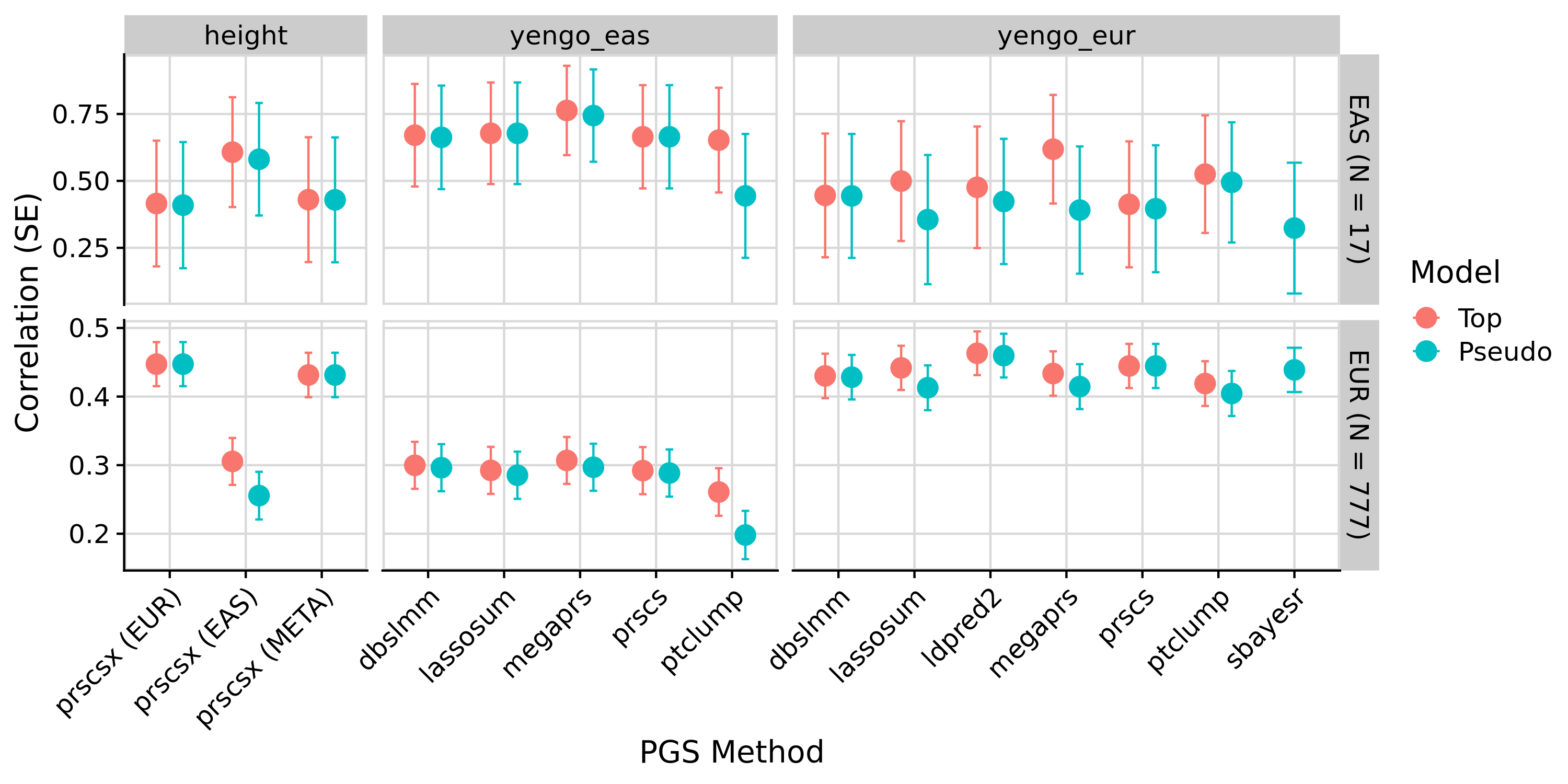
Yengo - EUR/EAS PGS correlation with height in OpenSNP EUR and EAS populations
Note. ‘external’ is based on a larger GWAS (incl. 23andMe)
GenoPred V1 comparison
We will run OpenSNP through GenoPred v1, to check whether updates have altered the output.
Show code
# Go to another version of the repo on CREATE
cd /users/k1806347/oliverpainfel/test/GenoPred
# Checkout to the v1 version of the repo
git checkout v1
Prepare input
Show code
library(data.table)
setwd('/users/k1806347/oliverpainfel/test/GenoPred/GenoPredPipe')
# target_list
target_list <- fread('target_list_example.txt')
target_list <- data.frame(
name = 'opensnp',
path='/users/k1806347/oliverpainfel/Data/OpenSNP/processed/geno/opensnp_train',
type = 'samp_imp_vcf',
output = '/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred_v1/test1',
indiv_report = F)
dir.create('misc/opensnp', recursive = T)
write.table(target_list, 'misc/opensnp/target_list.txt', col.names=T, row.names=F, quote=F, sep=' ')
# gwas_list
yengo_eur <- fread('/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eur.txt')
yengo_eur <- yengo_eur[, c('variant_id','effect_allele','other_allele','beta','standard_error','effect_allele_frequency','p_value','n'), with=F]
names(yengo_eur) <- c('SNP','A1','A2','BETA','SE','FREQ','P','N')
fwrite(yengo_eur, '/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eur.format.txt', sep = ' ', quote = F, na = 'NA')
yengo_eas <- fread('/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eas.txt')
yengo_eas <- yengo_eas[, c('variant_id','effect_allele','other_allele','beta','standard_error','effect_allele_frequency','p_value','n'), with=F]
names(yengo_eas) <- c('SNP','A1','A2','BETA','SE','FREQ','P','N')
fwrite(yengo_eas, '/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eas.format.txt', sep = ' ', quote = F, na = 'NA')
gwas_list <- fread('gwas_list_example.txt')
gwas_list<-rbind(gwas_list, data.table(
name='yengoeur',
path='/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eur.format.txt',
population='EUR',
sampling=NA,
prevalence=NA,
mean=NA,
sd=NA,
label="\"Yengo 2022 Height EUR\""))
gwas_list<-rbind(gwas_list, data.table(
name='yengoeas',
path='/users/k1806347/oliverpainfel/Data/GWAS_sumstats/opensnp_test/yengo_2022_height_eas.format.txt',
population='EAS',
sampling=NA,
prevalence=NA,
mean=NA,
sd=NA,
label="\"Yengo 2022 Height EAS\""))
gwas_list<-gwas_list[gwas_list$name %in% c('yengoeur','yengoeas'),]
write.table(gwas_list, 'misc/opensnp/gwas_list.txt', col.names=T, row.names=F, quote=F, sep=' ')
# score_list
score_list <- fread('score_list_example.txt')
score_list <- score_list[-1,]
write.table(score_list, 'misc/opensnp/score_list.txt', col.names=T, row.names=F, quote=F, sep=' ')
# config
config <- c(
"gwas_list: misc/opensnp/gwas_list.txt",
"target_list: misc/opensnp/target_list.txt",
"score_list: misc/opensnp/score_list.txt"
)
write.table(config, 'misc/opensnp/config.yaml', col.names=F, row.names=F, quote=F, sep=' ')Run GenoPred v1
Show code
# Calculate score using all methods
snakemake -n --profile slurm --configfile=misc/opensnp/config.yaml --use-conda run_target_prs_all
Evaluate PGS
Show code
# Test correlation between PGS and phenotype
setwd('/scratch//prj/oliverpainfel/test/GenoPred/pipeline/')
library(data.table)
library(Hmisc)
# Read in pheno data
pheno <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/processed/pheno/height.txt')
pheno$FID<-0
# Read in ancestry data
keep_list <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred/test1/opensnp/ancestry/keep_list.txt')
# Read in pgs
gwas_list <- fread('misc/opensnp/gwas_list.txt')
gwas_list$name<-gsub('_','',gwas_list$name)
pgs_methods <- c('pt_clump','dbslmm','prscs','sbayesr','lassosum','ldpred2','megaprs')
pgs_methods_eur <- c('pt_clump','lassosum','megaprs')
pgs <- list()
for(pop_i in keep_list$POP){
pgs[[pop_i]] <- list()
for(gwas_i in gwas_list$name){
pgs[[pop_i]][[gwas_i]] <- list()
for(pgs_method_i in pgs_methods){
if(gwas_list$population[gwas_list$name == gwas_i] == 'EUR' | (gwas_list$population[gwas_list$name == gwas_i] != 'EUR' & (pgs_method_i %in% pgs_methods_eur))){
pgs[[pop_i]][[gwas_i]][[pgs_method_i]] <- fread(paste0('/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred_v1/test1/opensnp/prs/',pop_i,'/',pgs_method_i,'/',gwas_i,'/opensnp.',gwas_i,'.',pop_i,'.profiles'))
}
}
}
}
# Estimate correlation between pheno and pgs
cor <- NULL
for(pop_i in names(pgs)){
for(gwas_i in names(pgs[[pop_i]])){
for(pgs_method_i in names(pgs[[pop_i]][[gwas_i]])){
pgs_i <- pgs[[pop_i]][[gwas_i]][[pgs_method_i]]
pheno_pgs<-merge(pheno, pgs_i, by = c('FID','IID'))
for(model_i in names(pgs_i)[-1:-2]){
y <- scale(pheno_pgs$height)
x <- scale(pheno_pgs[[model_i]])
if(all(is.na(x))){
next
}
coef_i <- coef(summary(mod <- lm(y ~ x)))
tmp <- data.table(
pop = pop_i,
gwas = gwas_i,
pgs_method = pgs_method_i,
name = model_i,
r = coef_i[2,1],
se = coef_i[2,2],
p = coef_i[2,4],
n = nobs(mod))
cor <- rbind(cor, tmp)
}
}
}
}
# Save the results
dir.create('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc')
write.csv(
cor,
'/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-v1-yengo-assoc.csv',
row.names = F
)
cor<-fread('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-v1-yengo-assoc.csv')
# Restrict to best only
cor_subset <- NULL
for(pop_i in unique(cor$pop)){
for(gwas_i in unique(cor$gwas[cor$pop == pop_i])){
for(pgs_method_i in unique(cor$pgs_method[cor$pop == pop_i & cor$gwas == gwas_i])){
# Subset relevant results
cor_i <- cor[
cor$pop == pop_i &
cor$gwas == gwas_i &
cor$pgs_method == pgs_method_i,]
# Top R
top_i <- cor_i[which(cor_i$r == max(cor_i$r, na.rm = T))[1],]
cor_subset <- rbind(cor_subset, top_i)
}
}
}
# Read in the associations for the best PGS when using the new version of GenoPred
cor_new <- fread('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-yengo-assoc.csv')
cor_new_subset <- NULL
for(pop_i in unique(cor_new$pop)){
for(gwas_i in unique(cor_new$gwas[cor_new$pop == pop_i])){
for(pgs_method_i in unique(cor_new$pgs_method[cor_new$pop == pop_i & cor_new$gwas == gwas_i])){
# Subset relevant results
cor_new_i <- cor_new[
cor_new$pop == pop_i &
cor_new$gwas == gwas_i &
cor_new$pgs_method == pgs_method_i,]
# Top R
top_i <- cor_new_i[which(cor_new_i$r == max(cor_new_i$r, na.rm = T))[1],]
cor_new_subset <- rbind(cor_new_subset, top_i)
}
}
}
# Compare the results
cor_subset$Version <- 'GenoPred V1'
cor_subset$pgs_method<-gsub('_', '', cor_subset$pgs_method)
cor_new_subset$Version <- 'GenoPred V2'
cor_new_subset$gwas<-gsub('_', '', cor_new_subset$gwas)
cor_both <- rbind(cor_subset, cor_new_subset)
cor_both$pop <- paste0(cor_both$pop, "\n N = ", cor_both$n)
##
# Plot the results
##
# yengo_eur
tmp <- cor_both[cor_both$gwas == 'yengoeur',]
y_lim <- c(min(tmp$r - tmp$se), max(tmp$r + tmp$se))
v1_plot <-
ggplot(cor_both[cor_both$gwas == 'yengoeur' & cor_both$Version == 'GenoPred V1', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.7, fill = '#F8766D') +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(pop ~ Version) +
panel_border()
v2_plot <-
ggplot(cor_both[cor_both$gwas == 'yengoeur' & cor_both$Version == 'GenoPred V2', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.7, fill = '#00BFC4') +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(pop ~ Version) +
panel_border()
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-v1-comp-yengo_eur.png',
units = 'px',
width = 3000,
height = 3000,
res = 300)
plot_grid(v1_plot, v2_plot, labels = NULL)
dev.off()
###
# Compare again using same ancestry classification threshold
###
# Read in ancestry predictions
model_pred <- fread('/users/k1806347/oliverpainfel/Data/OpenSNP/GenoPred_v1/test1/opensnp/ancestry/ancestry_all/opensnp.Ancestry.model_pred')
pgs_strict <- list()
for(pop_i in keep_list$POP){
pop_i_keep <- model_pred[model_pred[[pop_i]] > 0.95, ]
pgs_strict[[pop_i]] <- list()
for(gwas_i in gwas_list$name){
pgs_strict[[pop_i]][[gwas_i]] <- list()
for(pgs_method_i in pgs_methods){
if(gwas_list$population[gwas_list$name == gwas_i] == 'EUR' | (gwas_list$population[gwas_list$name == gwas_i] != 'EUR' & (pgs_method_i %in% pgs_methods_eur))){
pgs_strict[[pop_i]][[gwas_i]][[pgs_method_i]] <- merge(pgs[[pop_i]][[gwas_i]][[pgs_method_i]], pop_i_keep[, c('FID','IID'), with = F], by = c('FID','IID'))
}
}
}
}
# Estimate correlation between pheno and pgs
cor <- NULL
for(pop_i in names(pgs_strict)){
for(gwas_i in names(pgs_strict[[pop_i]])){
for(pgs_strict_method_i in names(pgs_strict[[pop_i]][[gwas_i]])){
pgs_strict_i <- pgs_strict[[pop_i]][[gwas_i]][[pgs_strict_method_i]]
pheno_pgs_strict<-merge(pheno, pgs_strict_i, by = c('FID','IID'))
for(model_i in names(pgs_strict_i)[-1:-2]){
y <- scale(pheno_pgs_strict$height)
x <- scale(pheno_pgs_strict[[model_i]])
if(all(is.na(x))){
next
}
coef_i <- coef(summary(mod <- lm(y ~ x)))
tmp <- data.table(
pop = pop_i,
gwas = gwas_i,
pgs_method = pgs_strict_method_i,
name = model_i,
r = coef_i[2,1],
se = coef_i[2,2],
p = coef_i[2,4],
n = nobs(mod))
cor <- rbind(cor, tmp)
}
}
}
}
# Save the results
dir.create('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc')
write.csv(
cor,
'/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-v1-strict-yengo-assoc.csv',
row.names = F
)
cor<-fread('/scratch/prj/oliverpainfel/Data/OpenSNP/assoc/genopred-v1-strict-yengo-assoc.csv')
# Restrict to best only
cor_subset <- NULL
for(pop_i in unique(cor$pop)){
for(gwas_i in unique(cor$gwas[cor$pop == pop_i])){
for(pgs_method_i in unique(cor$pgs_method[cor$pop == pop_i & cor$gwas == gwas_i])){
# Subset relevant results
cor_i <- cor[
cor$pop == pop_i &
cor$gwas == gwas_i &
cor$pgs_method == pgs_method_i,]
# Top R
top_i <- cor_i[which(cor_i$r == max(cor_i$r, na.rm = T))[1],]
cor_subset <- rbind(cor_subset, top_i)
}
}
}
# Compare the results
cor_subset$Version <- 'GenoPred V1'
cor_subset$pgs_method<-gsub('_', '', cor_subset$pgs_method)
cor_new_subset$Version <- 'GenoPred V2'
cor_new_subset$gwas<-gsub('_', '', cor_new_subset$gwas)
cor_both <- rbind(cor_subset, cor_new_subset)
cor_both$pop <- paste0(cor_both$pop, "\n N = ", cor_both$n)
##
# Plot the results
##
# yengo_eur
tmp <- cor_both[cor_both$gwas == 'yengoeur',]
y_lim <- c(min(tmp$r - tmp$se), max(tmp$r + tmp$se))
v1_plot <-
ggplot(cor_both[cor_both$gwas == 'yengoeur' & cor_both$Version == 'GenoPred V1', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.7, fill = '#F8766D') +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(pop ~ Version) +
panel_border()
v2_plot <-
ggplot(cor_both[cor_both$gwas == 'yengoeur' & cor_both$Version == 'GenoPred V2', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.7, fill = '#00BFC4') +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(pop ~ Version) +
panel_border()
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-v1-comp_strict-yengo_eur.png',
units = 'px',
width = 3000,
height = 3000,
res = 300)
plot_grid(v1_plot, v2_plot, labels = NULL)
dev.off()
######
# Plot the same plot only using the EUR target population in OpenSNP
######
tmp <- cor_both[cor_both$gwas == 'yengoeur' & grepl('EUR', cor_both$pop), ]
y_lim <- c(min(tmp$r - tmp$se), max(tmp$r + tmp$se))
v1_plot <-
ggplot(tmp[tmp$Version == 'GenoPred V1', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
geom_point(stat = "identity", position = position_dodge(), size = 5, colour = '#F8766D') +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(. ~ Version) +
panel_border()
v2_plot <-
ggplot(tmp[tmp$Version == 'GenoPred V2', ],
aes(x = pgs_method, y = r)) +
geom_hline(yintercept = 0) +
geom_errorbar(
aes(ymin = r - se, ymax = r + se),
width = .2,
position = position_dodge(width = 0.7)
) +
geom_point(stat = "identity", position = position_dodge(), size = 5, colour = '#00BFC4') +
labs(
y = "Correlation (SE)",
x = 'PGS Method',
title = 'Yengo - EUR vs OpenSNP'
) +
ylim(y_lim) +
theme_half_open() +
background_grid() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5, size=12)) +
facet_grid(. ~ Version) +
panel_border()
png('/users/k1806347/oliverpainfel/Software/MyGit/GenoPred/docs/Images/OpenSNP/genopred-v1-comp_strict_eur-yengo_eur.png',
units = 'px',
width = 3000,
height = 1500,
res = 300)
plot_grid(v1_plot, v2_plot, labels = NULL)
dev.off()Show comparison results
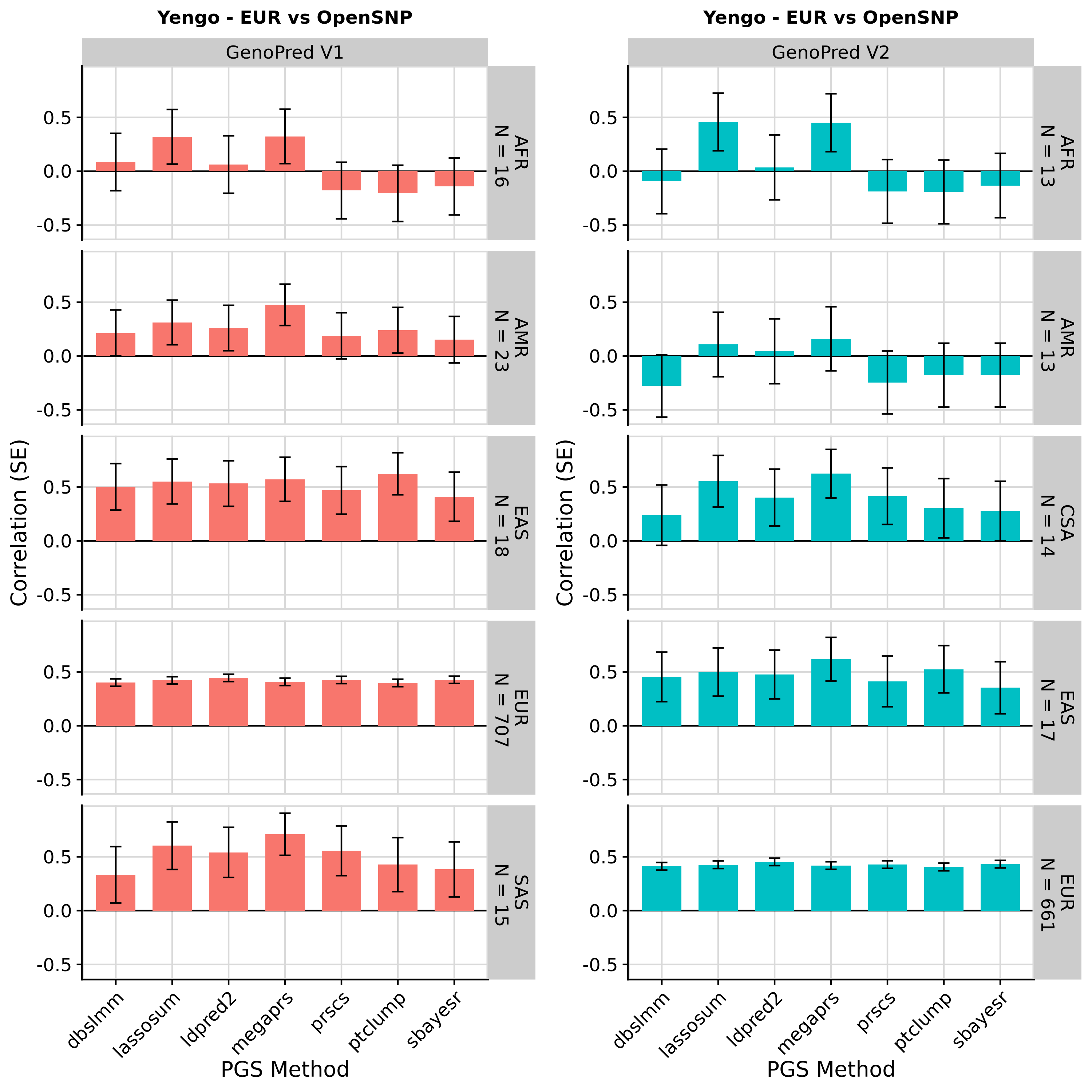
Out-of-the-box comparison
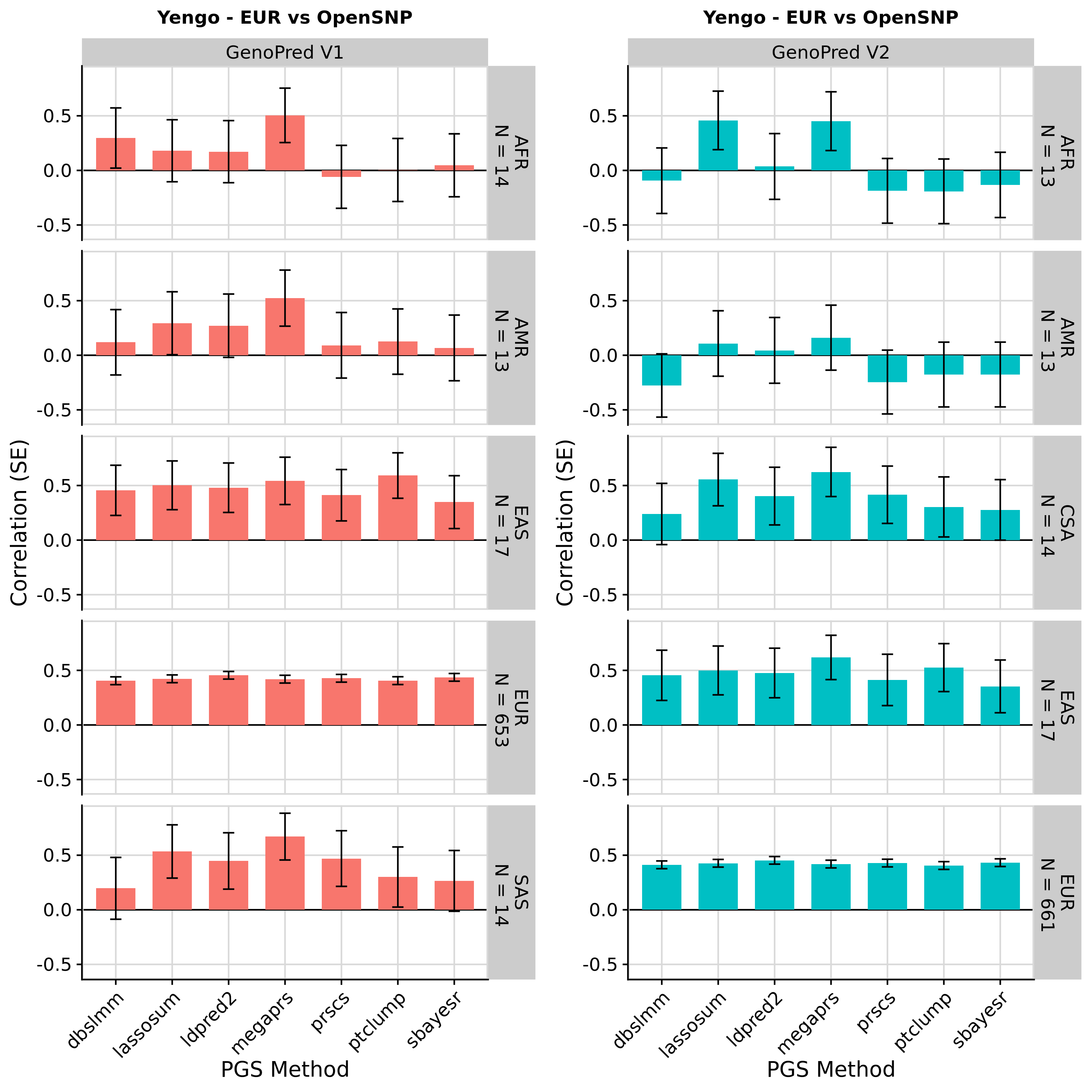
Using same ancestry classification threshold
Showing results in European OpenSNP data only
Conclusion
These results are as expected. PGS associations with height in OpenSNP are highly concordant across version of GenoPred. Small differences occur due to a more stringent ancestry classification threshold in GenoPred v2, typically increasing the correlation between the PGS and observed height. The externally derived score for height downloaded from the PGS Catalogue outperforms score created by GenoPred - This occurs because GenoPred used the publicly available GWAS summary statistics, exlcuding the 23andMe dataset, whereas the PGS weights downloaded from the PGS catalogue were derived using private GWAS sumstats, including 23andMe.